DMR detection with Methyl-IT
Analysis for Arabidopsis MSH1-memory versus non-memory first generation
Robersy Sanchez
rus547@psu.edu
Mackenzie’s lab
Department of Biology and Plant Science.
Pennsylvania State University, University Park, PA 16802
Maintainer: Robersy Sanchez
rus547@psu.edu
Mackenzie’s lab
Department of Biology and Plant Science.
Pennsylvania State University, University Park, PA 16802
Maintainer: Robersy Sanchez
15 January 2024
Source:vignettes/dmr_at_memory.Rmd
dmr_at_memory.RmdAbstract
Herein, an example of Methyl-IT application to the detection of differential methylated regions (DMRs) on first generation MSH1 RNAi transgene segregants data set is presented.
Introduction
MethylIT is an R package for methylome analysis based on a signal-detection machine learning approach (SD-ML). This approach is postulated to provide greater sensitivity for resolving true signal from the methylation background within the methylome (1, 2). Because the biological signal created within the dynamic methylome environment characteristic of plants is not free from background noise, the approach, designated Methyl-IT, includes the application of signal detection theory (3–6).
A basic requirement for the application of signal detection is the knowledge of the probability distribution of the background noise, which is used as null hypothesis in the detection of the methylation signal. This probability distribution is, in general, a member of generalized gamma distribution family, and it can be deduced on a statistical mechanical/thermodynamics basis from DNA methylation induced by thermal fluctuations (1).
Herein, we provide an example of Methyl-IT application to DMR estimation on patients with PALL (7). Due to the size of human methylome the current example only covers the analysis of chromosome 9. A full description of Methyl-IT application of methylome analysis for Clinical Diagnostics is given in the manuscript (2). A wider analysis of PALL data set is given in (8).
The first step of DMR estimation follows the general step of Methyl-IT pipeline for the estimation of DMPs. In Methyl-IT, a DMP is a cytosine positions that, with high probability, carries a statistically significant methylation change in respect to the same position in a control population. Once DMPs are identified, a simple algorithm can be applied to identify clusters of DMPs using dmpClusters function. Next, countTest2 is applied to test whether a given cluster of DMPs is a DMR.
It is worthy to notices that any clustering algorithm will impose non-natural constraints on the methylation patterning, which not necessarily will be associated to the local DNA physico-chemical properties and to local molecular evolutionary features of every DMR detected. Every cytosine methylation has a concrete physical effect of the DNA, altering the DNA persistence length, i.e., altering the local DNA flexibility (9–11). DNA flexibility was shown to affect the binding of proteins to methylated DNA and DNA-containing lesion (12, 13) and nucleosome mechanical stability (14). In other words, the sizes of naturally occurrying methylation motifs are not arbitrarily set, but very well constrained under molecular evolutionary pressure, probably, particular for each species.
The R packages required in this example are:
Estimation of differentially methylated regions (DMRs)
DMP data set is available for download.
## ===== Download methylation data from Github ========
url <- paste0("https://github.com/genomaths/genomaths.github.io/raw/master/AT_memory/",
"dmps_mm-vs-nm_all-context_gen1_nmRef_min-meth-3_cov-4_04-16-20.RData")
temp <- tempfile(fileext = ".RData")
download.file(url = url, destfile = temp)
load(temp)
file.remove(temp); rm(temp, url)The gene annotation
## ===== Download methylation data from GitHub ========
url <- paste0("https://github.com/genomaths/genomaths.github.io/raw/master/",
"Annotation/Arabidopsis_thaliana_genes_TAIR10.38.gtf.RData")
temp <- tempfile(fileext = ".RData")
download.file(url = url, destfile = temp)
load(temp)
file.remove(temp); rm(temp, url);
GENES <- genes[genes$type == "gene"]
## To get exons and introns
exons <- genes[genes$type == "exon"]
introns <- setdiff(x = genes, y = exons, ignore.strand = TRUE)
## To add the corresponding gene names to introns
hits <- findOverlaps(introns, genes, type = "within", ignore.strand = TRUE)
introns <- introns[queryHits(hits)]
mcols(introns) <- mcols(genes[subjectHits(hits)])Cluster of DMPs
Function dmpClusters search for cluster of DMPs found from all the samples. The basic idea reside in that if a given cluster of DMP is a DMR, then it will be consistently differentially methylated in all treatment samples, and we can identify them applying function countTest2.
There is not a unique approach to build DMP clusters, since several clustering criteria can be applied. Herein, we use the two approach implemented in dmpClusters function (follow the link or type ?dmpClusters to see the help for the function).
clust_50_100 = dmpClusters(GR = dmps, maxDist = 50, minNumDMPs = 10,
maxClustDist = 100, method = "fixed.int",
num.cores = 1L, verbose = FALSE)
clust_50_100## GRanges object with 18035 ranges and 1 metadata column:
## seqnames ranges strand | dmps
## <Rle> <IRanges> <Rle> | <integer>
## [1] 1 183-330 * | 27
## [2] 1 647-809 * | 10
## [3] 1 18052-18224 * | 14
## [4] 1 18643-18746 * | 12
## [5] 1 42279-42423 * | 40
## ... ... ... ... . ...
## [18031] 5 26670460-26670651 * | 15
## [18032] 5 26788467-26788575 * | 11
## [18033] 5 26945319-26945446 * | 11
## [18034] 5 26974071-26974175 * | 10
## [18035] 5 26974820-26975362 * | 39
## -------
## seqinfo: 5 sequences from an unspecified genome; no seqlengthsDMPs clusterization strongly depends on the maximum distance
permitted between two consecutive DMPs that belong to the same cluster
(parameter: minNumDMPs) and on the minimum number of DMPs
that must be present on a detected agglomeration of DMPs to be
considered a cluster (parameter: maxDist).
.clust_50_100 = dmpClusters(GR = dmps, maxDist = 50, minNumDMPs = 10,
maxClustDist = 100, method = "relaxed",
num.cores = 1L, verbose = FALSE)
.clust_50_100## GRanges object with 17528 ranges and 1 metadata column:
## seqnames ranges strand | dmps
## <Rle> <IRanges> <Rle> | <integer>
## [1] 1 94-330 * | 29
## [2] 1 647-880 * | 13
## [3] 1 17833-18512 * | 35
## [4] 1 18643-18746 * | 12
## [5] 1 42279-42423 * | 40
## ... ... ... ... . ...
## [17524] 5 26788399-26788575 * | 12
## [17525] 5 26835058-26835302 * | 13
## [17526] 5 26945319-26945446 * | 11
## [17527] 5 26974071-26974329 * | 14
## [17528] 5 26974728-26975362 * | 40
## -------
## seqinfo: 5 sequences from an unspecified genome; no seqlengthsAlternatively, we can use function dmpClustering from MethylIT.utils R package. Notice that although ‘method’ parameter are named the same, on each function these stand for different approaches.
clust.50.100 = dmpClustering(dmps = dmps, minNumDMPs = 10, win.size = 50,
step.size = 50, method = "fixed.int",
maxClustDist = 100, verbose = FALSE)
clust.50.100## GRanges object with 15521 ranges and 1 metadata column:
## seqnames ranges strand | dmps
## <Rle> <IRanges> <Rle> | <integer>
## [1] 1 94-330 * | 29
## [2] 1 647-880 * | 13
## [3] 1 17833-18746 * | 47
## [4] 1 42279-42423 * | 40
## [5] 1 44603-44848 * | 19
## ... ... ... ... . ...
## [15517] 5 26835058-26835302 * | 13
## [15518] 5 26891628-26892012 * | 10
## [15519] 5 26945319-26945446 * | 11
## [15520] 5 26974071-26974329 * | 14
## [15521] 5 26974728-26975362 * | 40
## -------
## seqinfo: 5 sequences from an unspecified genome; no seqlengths
.clust.50.100 = dmpClustering(dmps = dmps, minNumDMPs = 10, method = "relaxed",
maxClustDist = 100, verbose = FALSE)
.clust.50.100## GRanges object with 17647 ranges and 1 metadata column:
## seqnames ranges strand | dmps
## <Rle> <IRanges> <Rle> | <integer>
## [1] 1 94-330 * | 29
## [2] 1 647-880 * | 13
## [3] 1 17833-18512 * | 35
## [4] 1 18643-18746 * | 12
## [5] 1 42279-42423 * | 40
## ... ... ... ... . ...
## [17643] 5 26788399-26788575 * | 12
## [17644] 5 26835058-26835302 * | 13
## [17645] 5 26945319-26945446 * | 11
## [17646] 5 26974071-26974329 * | 14
## [17647] 5 26974728-26975362 * | 40
## -------
## seqinfo: 5 sequences from an unspecified genome; no seqlengthsDMR detection
The count of the number of DMPs on each region will be accomplished with getDMPatRegions function. This function operates based on findOverlaps function from the ‘GenomicRanges’ Bioconductor R package. The findOverlaps function has several critical parameters like, for example, ‘maxgap’, ‘minoverlap’, and ‘ignore.strand’. In our getDMPatRegions function, except for setting ignore.strand = TRUE and type = “within”, we preserve the rest of default ‘findOverlaps’ parameters.
Experiment design for each clustering approach
Design for method = “fixed.int” from dmpClusters function
## ==== Setting up the experiment design to test for DMRs ===
nams <- names(dmps)
dmps_at_clust_50_100 <- getDMPatRegions(GR = dmps, regions = clust_50_100,
ignore.strand = TRUE)
dmps_at_clust_50_100 <- uniqueGRanges(dmps_at_clust_50_100, columns = 1L,
ignore.strand = TRUE, type = 'equal',
verbose = FALSE)
colnames(mcols(dmps_at_clust_50_100)) <- nams
colData <- data.frame(condition = factor(c("MM", "MM","MM","MM", "MM",
"NM","NM","NM","NM"),
levels = c("NM", "MM")),
nams,
row.names = 2)
## Build a RangedGlmDataSet object
ds_50_100 <- glmDataSet(GR = dmps_at_clust_50_100, colData = colData)The GRanges object with DMPs on it
dmps_at_clust_50_100## GRanges object with 18035 ranges and 9 metadata columns:
## seqnames ranges strand | M_1_105 M_1_168 M_1_179
## <Rle> <IRanges> <Rle> | <numeric> <numeric> <numeric>
## [1] 1 183-330 * | 0 22 2
## [2] 1 647-809 * | 0 3 4
## [3] 1 18052-18224 * | 2 2 1
## [4] 1 18643-18746 * | 2 1 0
## [5] 1 42279-42423 * | 15 16 0
## ... ... ... ... . ... ... ...
## [18031] 5 26670460-26670651 * | 5 1 8
## [18032] 5 26788467-26788575 * | 7 1 1
## [18033] 5 26945319-26945446 * | 3 2 0
## [18034] 5 26974071-26974175 * | 3 4 0
## [18035] 5 26974820-26975362 * | 4 11 8
## M_1_27 M_1_3 NM_1_12 NM_1_45 NM_1_70 NM_1_86
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## [1] 3 1 0 0 2 0
## [2] 2 1 0 0 0 0
## [3] 3 7 0 0 0 0
## [4] 3 0 0 2 1 3
## [5] 13 27 0 0 0 0
## ... ... ... ... ... ... ...
## [18031] 3 0 0 0 0 0
## [18032] 1 0 1 0 0 0
## [18033] 2 2 2 1 0 1
## [18034] 3 0 1 4 2 3
## [18035] 3 11 2 3 0 0
## -------
## seqinfo: 5 sequences from an unspecified genome; no seqlengthsThe glmDataSet object.
ds_50_100## RangedGlmDataSet with 18035 regions and 9 columns (individuals) with factor levels 'NM' and 'MM'
## The accessible objects in the dataset are:
## Length Class Mode
## GR 18035 GRanges S4
## counts 162315 -none- numeric
## colData 1 data.frame list
## sampleNames 9 -none- character
## levels 2 -none- character
## optionData 0 -none- NULLDesign for method = “relaxed” from dmpClusters function
## ==== Setting up the experiment design to test for DMRs ===
nams <- names(dmps)
.dmps_at_clust_50_100 <- getDMPatRegions(GR = dmps, regions = .clust_50_100,
ignore.strand = TRUE)
.dmps_at_clust_50_100 <- uniqueGRanges(.dmps_at_clust_50_100, columns = 1L,
ignore.strand = TRUE, type = 'equal',
verbose = FALSE)
colnames(mcols(.dmps_at_clust_50_100)) <- nams
## Build a RangedGlmDataSet object
.ds_50_100 <- glmDataSet(GR = .dmps_at_clust_50_100, colData = colData)Design for method = “fixed.int” from dmpClustering function
## ==== Setting up the experiment design to test for DMRs ===
nams <- names(dmps)
dmps_at_clust.50.100 <- getDMPatRegions(GR = dmps, regions = clust.50.100,
ignore.strand = TRUE)
dmps_at_clust.50.100 <- uniqueGRanges(dmps_at_clust.50.100, columns = 1L,
ignore.strand = TRUE, type = 'equal',
verbose = FALSE)
colnames(mcols(dmps_at_clust.50.100)) <- nams
## Build a RangedGlmDataSet object
ds.50.100 <- glmDataSet(GR = dmps_at_clust.50.100, colData = colData)Design for method = “relaxed” from dmpClustering function
## ==== Setting up the experiment design to test for DMRs ===
nams <- names(dmps)
dmps_at.clust.50.100 <- getDMPatRegions(GR = dmps, regions = .clust.50.100,
ignore.strand = TRUE)
dmps_at.clust.50.100 <- uniqueGRanges(dmps_at.clust.50.100, columns = 1L,
ignore.strand = TRUE, type = 'equal',
verbose = FALSE)
colnames(mcols(dmps_at.clust.50.100)) <- nams
## Build a RangedGlmDataSet object
ds_50.100 <- glmDataSet(GR = dmps_at.clust.50.100, colData = colData)DMR detection
Herein, we are requesting a minimum density DMP of 1/100 bp = 0.01/bp (CountPerBp = 0.01), minimum of logarithm of fold changes in the number of DMPs, treatment versus control samples, equal to 1 (Minlog2FC = 1). Also, it is requested a coefficient of variation, on each sample set, not greater than 1 (maxGrpCV = c(1, 1)).
Notice that in the case of species with big genes, for example, human genes, we might want to use lower densities, e.g., CountPerBp = 0.001.
DMRs derived method = “fixed.int” from dmpClusters function
dmrs_fixed.int = countTest2(DS = ds_50_100,
minCountPerIndv = 8,
maxGrpCV = c(1, 1),
Minlog2FC = 1,
CountPerBp = 0.01,
test = 'LRT',
num.cores = 4L,
verbose = TRUE)
dmrs_fixed.intThe DMR associated genes at a \(distance \leq 1000\):
hits <- distanceToNearest(dmrs_fixed.int, GENES, ignore.strand=FALSE)
## DMR associated genes
hits <- hits[mcols(hits)[,1] <= 1000]
GENES[subjectHits(hits)]## GRanges object with 975 ranges and 4 metadata columns:
## seqnames ranges strand | gene_id type gene_biotype
## <Rle> <IRanges> <Rle> | <character> <factor> <character>
## [1] 1 43087-43295 - | AT1G04003 gene lncRNA
## [2] 1 239841-242703 - | AT1G01660 gene protein_coding
## [3] 1 258717-258963 - | AT1G04037 gene lncRNA
## [4] 1 428650-430720 - | AT1G02220 gene protein_coding
## [5] 1 1194187-1196889 + | AT1G04425 gene ncRNA
## ... ... ... ... . ... ... ...
## [971] 5 23775891-23779855 + | AT5G58880 gene protein_coding
## [972] 5 25934568-25935612 + | AT5G64890 gene protein_coding
## [973] 5 25967824-25968638 + | AT5G65005 gene protein_coding
## [974] 5 26068112-26069652 + | AT5G65230 gene protein_coding
## [975] 5 26411796-26414609 - | AT5G66050 gene protein_coding
## gene_name
## <character>
## [1] <NA>
## [2] <NA>
## [3] <NA>
## [4] NAC003
## [5] <NA>
## ... ...
## [971] <NA>
## [972] PEP2
## [973] <NA>
## [974] AtMYB53
## [975] <NA>
## -------
## seqinfo: 5 sequences from an unspecified genome; no seqlengthsDMRs derived method = “relaxed” from dmpClusters function
.dmrs_relaxed = countTest2(DS = .ds_50_100,
minCountPerIndv = 8,
maxGrpCV = c(1, 1),
Minlog2FC = 1,
CountPerBp = 0.01,
test = 'LRT',
num.cores = 4L,
verbose = TRUE)
.dmrs_relaxedThe DMR assciated genes at a \(distance \leq 1000\):
hits <- distanceToNearest(.dmrs_relaxed, GENES, ignore.strand=FALSE)
## DMR associated genes
hits <- hits[mcols(hits)[,1] <= 1000]
GENES[subjectHits(hits)]## GRanges object with 1215 ranges and 4 metadata columns:
## seqnames ranges strand | gene_id type
## <Rle> <IRanges> <Rle> | <character> <factor>
## [1] 1 43087-43295 - | AT1G04003 gene
## [2] 1 239841-242703 - | AT1G01660 gene
## [3] 1 258717-258963 - | AT1G04037 gene
## [4] 1 428650-430720 - | AT1G02220 gene
## [5] 1 1194187-1196889 + | AT1G04425 gene
## ... ... ... ... . ... ...
## [1211] 5 25934568-25935612 + | AT5G64890 gene
## [1212] 5 25967824-25968638 + | AT5G65005 gene
## [1213] 5 26068112-26069652 + | AT5G65230 gene
## [1214] 5 26161624-26169191 - | AT5G65460 gene
## [1215] 5 26411796-26414609 - | AT5G66050 gene
## gene_biotype gene_name
## <character> <character>
## [1] lncRNA <NA>
## [2] protein_coding <NA>
## [3] lncRNA <NA>
## [4] protein_coding NAC003
## [5] ncRNA <NA>
## ... ... ...
## [1211] protein_coding PEP2
## [1212] protein_coding <NA>
## [1213] protein_coding AtMYB53
## [1214] protein_coding KCA2
## [1215] protein_coding <NA>
## -------
## seqinfo: 5 sequences from an unspecified genome; no seqlengthsDMRs derived method = “fixed.int” from dmpClustering function
dmrs.fixed.int = countTest2(DS = ds.50.100,
minCountPerIndv = 8,
maxGrpCV = c(1, 1),
Minlog2FC = 1,
CountPerBp = 0.01,
test = 'LRT',
num.cores = 4L,
verbose = TRUE)
dmrs.fixed.intThe DMR assciated genes at a \(distance \leq 1000\):
hits <- distanceToNearest(dmrs.fixed.int, GENES, ignore.strand=FALSE)
## DMR associated genes
hits <- hits[mcols(hits)[,1] <= 1000]
GENES[subjectHits(hits)]## GRanges object with 1151 ranges and 4 metadata columns:
## seqnames ranges strand | gene_id type
## <Rle> <IRanges> <Rle> | <character> <factor>
## [1] 1 43087-43295 - | AT1G04003 gene
## [2] 1 258717-258963 - | AT1G04037 gene
## [3] 1 428650-430720 - | AT1G02220 gene
## [4] 1 1194187-1196889 + | AT1G04425 gene
## [5] 1 1431177-1433010 - | AT1G05010 gene
## ... ... ... ... . ... ...
## [1147] 5 25934568-25935612 + | AT5G64890 gene
## [1148] 5 25967824-25968638 + | AT5G65005 gene
## [1149] 5 26068112-26069652 + | AT5G65230 gene
## [1150] 5 26161624-26169191 - | AT5G65460 gene
## [1151] 5 26411796-26414609 - | AT5G66050 gene
## gene_biotype gene_name
## <character> <character>
## [1] lncRNA <NA>
## [2] lncRNA <NA>
## [3] protein_coding NAC003
## [4] ncRNA <NA>
## [5] protein_coding ACO4
## ... ... ...
## [1147] protein_coding PEP2
## [1148] protein_coding <NA>
## [1149] protein_coding AtMYB53
## [1150] protein_coding KCA2
## [1151] protein_coding <NA>
## -------
## seqinfo: 5 sequences from an unspecified genome; no seqlengthsDMRs derived method = “relaxed” from dmpClustering function
dmrs.relaxed = countTest2(DS = ds_50.100,
minCountPerIndv = 8,
maxGrpCV = c(1, 1),
Minlog2FC = 1,
CountPerBp = 0.01,
test = 'LRT',
num.cores = 4L,
verbose = TRUE)
dmrs.relaxedThe DMR assciated genes at a \(distance \leq 1000\):
hits <- distanceToNearest(dmrs.relaxed, GENES, ignore.strand=FALSE)
## DMR associated genes
hits <- hits[mcols(hits)[,1] <= 1000]
GENES[subjectHits(hits)]## GRanges object with 1214 ranges and 4 metadata columns:
## seqnames ranges strand | gene_id type
## <Rle> <IRanges> <Rle> | <character> <factor>
## [1] 1 43087-43295 - | AT1G04003 gene
## [2] 1 239841-242703 - | AT1G01660 gene
## [3] 1 258717-258963 - | AT1G04037 gene
## [4] 1 428650-430720 - | AT1G02220 gene
## [5] 1 1194187-1196889 + | AT1G04425 gene
## ... ... ... ... . ... ...
## [1210] 5 25934568-25935612 + | AT5G64890 gene
## [1211] 5 25967824-25968638 + | AT5G65005 gene
## [1212] 5 26068112-26069652 + | AT5G65230 gene
## [1213] 5 26161624-26169191 - | AT5G65460 gene
## [1214] 5 26411796-26414609 - | AT5G66050 gene
## gene_biotype gene_name
## <character> <character>
## [1] lncRNA <NA>
## [2] protein_coding <NA>
## [3] lncRNA <NA>
## [4] protein_coding NAC003
## [5] ncRNA <NA>
## ... ... ...
## [1210] protein_coding PEP2
## [1211] protein_coding <NA>
## [1212] protein_coding AtMYB53
## [1213] protein_coding KCA2
## [1214] protein_coding <NA>
## -------
## seqinfo: 5 sequences from an unspecified genome; no seqlengthsNaturally ocurring regions
As was mentioned in the introduction algorithmically identified DMRs do not necessarily will be associated to the local DNA physico-chemical properties and to local molecular evolutionary features of every DMR detected.
All the weight of the evolutionary pressure lie down on naturally ocurrying methylation patternings inside, for example (but not limited to), exons and introns.
For introns we have:
## ==== Setting up the experiment design to test for DMRs ===
nams <- names(dmps)
dmps_at_exons <- getDIMPatGenes(GR = dmps, GENES = exons,
ignore.strand = TRUE,
output = "GRanges")
## Build a RangedGlmDataSet object
ds_exons <- glmDataSet(GR = dmps_at_exons, colData = colData)Differentially methylated exons
dmrs_exons = countTest2(DS = ds_exons,
minCountPerIndv = 8,
maxGrpCV = c(1, 1),
Minlog2FC = 1,
CountPerBp = 0.01,
test = 'LRT',
num.cores = 4L,
verbose = TRUE)## *** Number of GR after filtering counts 179## *** GLM...
dmrs_exons## GRanges object with 149 ranges and 17 metadata columns:
## seqnames ranges strand | M_1_105 M_1_168 M_1_179
## <Rle> <IRanges> <Rle> | <numeric> <numeric> <numeric>
## [1] 1 627482-628469 * | 0 0 30
## [2] 1 1506117-1509152 * | 42 0 44
## [3] 1 3572879-3573173 * | 5 7 26
## [4] 1 3892580-3894677 * | 24 17 23
## [5] 1 3979823-3981079 * | 25 9 13
## ... ... ... ... . ... ... ...
## [145] 5 19867969-19868877 * | 24 14 20
## [146] 5 21103008-21103568 * | 6 20 0
## [147] 5 21167432-21169462 * | 46 30 19
## [148] 5 22222886-22224526 * | 25 17 22
## [149] 5 22478712-22480498 * | 27 0 36
## M_1_27 M_1_3 NM_1_12 NM_1_45 NM_1_70 NM_1_86 log2FC
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## [1] 25 20 0 0 0 0 2.72579
## [2] 35 58 0 0 0 0 3.60550
## [3] 7 5 1 0 0 0 2.17475
## [4] 22 23 0 7 5 0 1.74047
## [5] 20 15 7 0 4 0 1.53471
## ... ... ... ... ... ... ... ...
## [145] 6 20 2 0 0 0 2.47373
## [146] 8 34 2 0 0 2 1.98787
## [147] 27 27 8 4 6 9 1.48497
## [148] 14 12 1 6 2 2 1.87877
## [149] 18 27 0 0 0 0 3.11795
## scaled.deviance pvalue model adj.pval CT.SignalDensity
## <numeric> <numeric> <character> <numeric> <numeric>
## [1] 86.6123 1.32022e-20 Neg.Binomial.W 7.86853e-20 0.000000000
## [2] 184.0775 6.24017e-42 Neg.Binomial 2.32446e-40 0.000000000
## [3] 17.4978 2.87643e-05 Neg.Binomial 3.86115e-05 0.000847458
## [4] 62.9822 2.08580e-15 Neg.Binomial 8.39957e-15 0.001429933
## [5] 23.1115 1.52876e-06 Neg.Binomial 2.42325e-06 0.002187749
## ... ... ... ... ... ...
## [145] 42.93430 5.66094e-11 Neg.Binomial 1.36045e-10 0.000550055
## [146] 9.76251 1.78107e-03 Neg.Binomial 1.90920e-03 0.001782531
## [147] 41.38165 1.25228e-10 Neg.Binomial 2.87060e-10 0.003323486
## [148] 45.37607 1.62609e-11 Neg.Binomial 4.32657e-11 0.001675807
## [149] 104.45853 1.60518e-24 Neg.Binomial 1.13892e-23 0.000000000
## TT.SignalDensity SignalDensityVariation
## <numeric> <numeric>
## [1] 0.0151822 0.01518219
## [2] 0.0117918 0.01179183
## [3] 0.0338983 0.03305085
## [4] 0.0103908 0.00896092
## [5] 0.0130469 0.01085919
## ... ... ...
## [145] 0.0184818 0.01793179
## [146] 0.0242424 0.02245989
## [147] 0.0146726 0.01134909
## [148] 0.0109689 0.00929311
## [149] 0.0120873 0.01208730
## -------
## seqinfo: 5 sequences from an unspecified genome; no seqlengthsFor introns we have:
## ==== Setting up the experiment design to test for DMRs ===
nams <- names(dmps)
dmps_at_introns <- getDIMPatGenes(GR = dmps, GENES = introns,
ignore.strand = TRUE,
output = "GRanges")
## Build a RangedGlmDataSet object
ds_introns <- glmDataSet(GR = dmps_at_introns, colData = colData)Differentially methylated introns
dmrs_introns = countTest2(DS = ds_introns,
minCountPerIndv = 8,
maxGrpCV = c(1, 1),
Minlog2FC = 1,
CountPerBp = 0.01,
test = 'LRT',
num.cores = 4L,
verbose = TRUE)## *** Number of GR after filtering counts 5## *** GLM...
dmrs_introns## GRanges object with 5 ranges and 17 metadata columns:
## seqnames ranges strand | M_1_105 M_1_168 M_1_179
## <Rle> <IRanges> <Rle> | <numeric> <numeric> <numeric>
## [1] 1 13148737-13149481 * | 14 14 6
## [2] 1 17534249-17534923 * | 6 14 7
## [3] 2 5885110-5885454 * | 4 12 10
## [4] 5 21161354-21162074 * | 0 0 10
## [5] 5 21174335-21175487 * | 12 19 13
## M_1_27 M_1_3 NM_1_12 NM_1_45 NM_1_70 NM_1_86 log2FC
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## [1] 10 10 0 0 0 0 2.47751
## [2] 11 7 2 0 0 2 1.60944
## [3] 5 9 5 1 1 1 1.38629
## [4] 23 10 0 0 2 0 1.85630
## [5] 10 9 0 2 0 3 1.79914
## scaled.deviance pvalue model adj.pval CT.SignalDensity
## <numeric> <numeric> <character> <numeric> <numeric>
## [1] 85.35744 2.49023e-20 Neg.Binomial.W 1.24512e-19 0.000000000
## [2] 25.21553 5.12678e-07 Neg.Binomial 8.54464e-07 0.001481481
## [3] 14.20990 1.63508e-04 Neg.Binomial 2.04385e-04 0.005797101
## [4] 6.92139 8.51706e-03 Neg.Binomial 8.51706e-03 0.000693481
## [5] 38.99278 4.25375e-10 Neg.Binomial 1.06344e-09 0.001084128
## TT.SignalDensity SignalDensityVariation
## <numeric> <numeric>
## [1] 0.0144966 0.01449664
## [2] 0.0133333 0.01185185
## [3] 0.0231884 0.01739130
## [4] 0.0119279 0.01123440
## [5] 0.0109280 0.00984389
## -------
## seqinfo: 3 sequences from an unspecified genome; no seqlengthsDifferentially methylated genes DMGs
The last results for introns and exons contrasts with the result for the whole gene-body.
## ==== Setting up the experiment design to test for DMRs ===
nams <- names(dmps)
dmps_at_genes <- getDIMPatGenes(GR = dmps, GENES = genes,
ignore.strand = TRUE,
output = "GRanges")
## Build a RangedGlmDataSet object
ds_genes <- glmDataSet(GR = dmps_at_genes, colData = colData)Differentically methylated genes
dmrs_genes = countTest2(DS = ds_genes,
minCountPerIndv = 8,
maxGrpCV = c(1, 1),
Minlog2FC = 1,
CountPerBp = 0.01,
test = 'LRT',
num.cores = 4L,
verbose = TRUE)## *** Number of GR after filtering counts 704## *** GLM...
dmrs_genes## GRanges object with 646 ranges and 17 metadata columns:
## seqnames ranges strand | M_1_105 M_1_168 M_1_179
## <Rle> <IRanges> <Rle> | <numeric> <numeric> <numeric>
## [1] 1 1444983-1446116 * | 11 15 12
## [2] 1 2157658-2158158 * | 8 10 18
## [3] 1 2993381-2993820 * | 10 6 24
## [4] 1 3572674-3573173 * | 10 14 52
## [5] 1 3892580-3894677 * | 49 37 48
## ... ... ... ... . ... ... ...
## [642] 5 21167432-21169462 * | 92 60 38
## [643] 5 21174321-21175857 * | 18 27 17
## [644] 5 22222886-22225616 * | 42 28 36
## [645] 5 23024896-23025915 * | 19 38 17
## [646] 5 24526245-24526636 * | 4 8 14
## M_1_27 M_1_3 NM_1_12 NM_1_45 NM_1_70 NM_1_86 log2FC
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## [1] 22 9 7 2 2 7 1.12059
## [2] 16 8 4 2 0 2 1.46634
## [3] 2 2 2 0 0 6 1.18377
## [4] 14 10 2 0 0 0 2.63906
## [5] 45 47 0 16 11 8 1.55571
## ... ... ... ... ... ... ... ...
## [642] 54 54 16 8 12 18 1.48497
## [643] 12 23 0 2 0 3 2.20460
## [644] 23 20 2 11 3 3 1.83636
## [645] 19 33 2 4 6 14 1.35504
## [646] 4 10 2 0 0 0 1.79176
## scaled.deviance pvalue model adj.pval CT.SignalDensity
## <numeric> <numeric> <character> <numeric> <numeric>
## [1] 16.07033 6.10329e-05 Neg.Binomial 7.76127e-05 0.00396825
## [2] 25.55214 4.30588e-07 Neg.Binomial 7.81348e-07 0.00399202
## [3] 4.41686 3.55854e-02 Neg.Binomial 3.55854e-02 0.00454545
## [4] 24.79981 6.36036e-07 Neg.Binomial 1.11956e-06 0.00100000
## [5] 111.81692 3.91877e-26 Neg.Binomial 7.67128e-25 0.00417064
## ... ... ... ... ... ...
## [642] 48.4620 3.36757e-12 Neg.Binomial 1.36821e-11 0.006646972
## [643] 63.4528 1.64254e-15 Neg.Binomial 9.55926e-15 0.000813273
## [644] 42.8798 5.82085e-11 Neg.Binomial 1.94833e-10 0.001739290
## [645] 18.3625 1.82615e-05 Neg.Binomial 2.58138e-05 0.006372549
## [646] 20.6233 5.59125e-06 Neg.Binomial 8.62040e-06 0.001275510
## TT.SignalDensity SignalDensityVariation
## <numeric> <numeric>
## [1] 0.0121693 0.00820106
## [2] 0.0239521 0.01996008
## [3] 0.0200000 0.01545455
## [4] 0.0400000 0.03900000
## [5] 0.0215443 0.01737369
## ... ... ...
## [642] 0.0293452 0.02269818
## [643] 0.0126220 0.01180872
## [644] 0.0109118 0.00917246
## [645] 0.0247059 0.01833333
## [646] 0.0204082 0.01913265
## -------
## seqinfo: 5 sequences from an unspecified genome; no seqlengthsIt seems to be that methylation patterning inside of gene-body forms a sort continuous regions simultaneously covering portions of exon and introns. In particular, this is true for genes experiencing alternative splicing, where some regions inside introns works as exons as well.
DMR finder function
Following MethylIT pipeline, DMRs can be identified in control and in treatment groups. The concept of DMR is subjective in some extend. That is, while MethylIT pipeline identify cytosine positions with high probability to be differential methylated in the treatment group with respect to the control group, it will correspond to the user to define whether to say, e.g., a region 100 bp long carrying 3 DMPs is a DMR. There is not theoretical ground to decide whether such a DMR would be biologically relevant or not for given species. Only the experimentation or further downstream analyses including, for example, the network enrichment analysis of DMR-associated genes would gives us an answer.
Herein, we propose third function to search for DMRs: dmrfinder
Let’s search for hypo-methylated DMRs with at least 20 DMPs on 200 bp. The graphics of samples 1 to 9 on chromosome “1” are returned.
hyp_dmrs <- dmrfinder(
obj = dmps,
hypo = TRUE,
min.signal = 10L,
win.size = 200,
step.size = 50,
stat = "count",
tv.colum = 8L,
naming = TRUE,
plots = TRUE,
mfcol = c(3, 3),
num.plot = 9,
verbose = FALSE)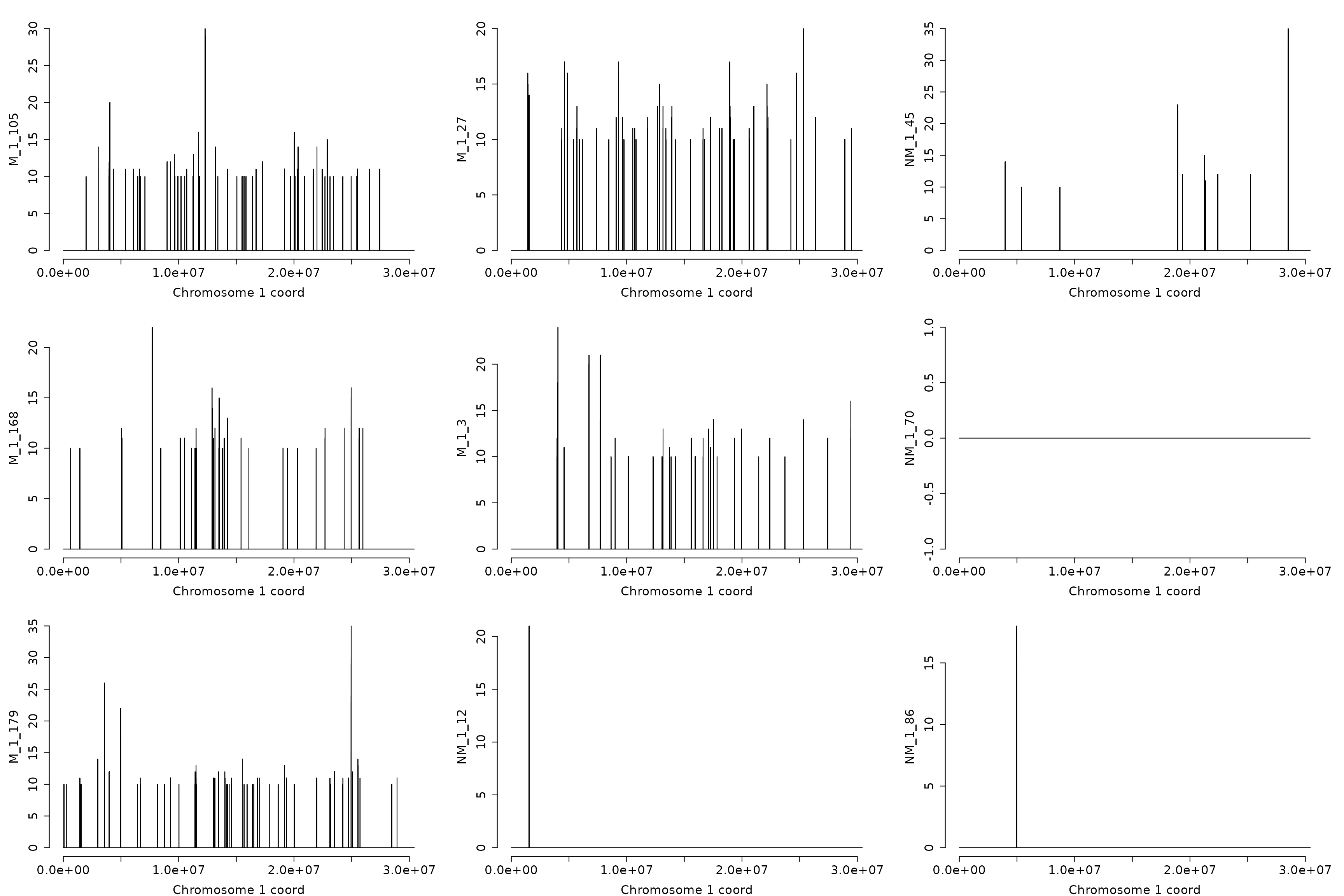
The hyper-DMRs on chromosome “1”
hyp_dmrs <- dmrfinder(
obj = dmps,
hyper = TRUE,
min.signal = 10L,
win.size = 200,
step.size = 50,
stat = "count",
tv.colum = 8L,
naming = TRUE,
plots = TRUE,
mfcol = c(3, 3),
num.plot = 9,
verbose = FALSE)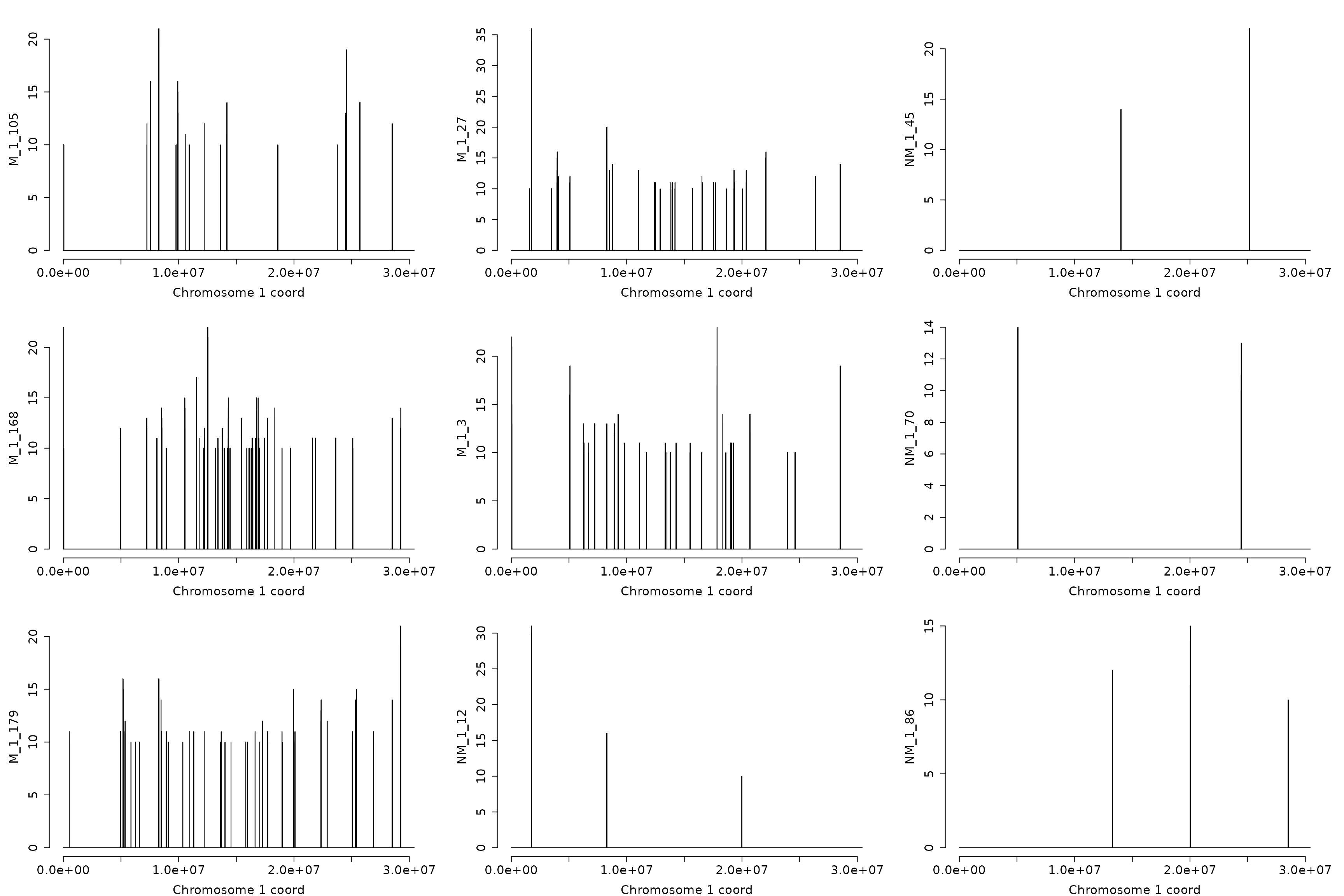
The hypo-DMRs on chromosome “2”
hyp_dmrs <- dmrfinder(
obj = dmps,
hyper = TRUE,
min.signal = 10L,
win.size = 200,
step.size = 50,
stat = "count",
tv.colum = 8L,
naming = TRUE,
plots = TRUE,
mfcol = c(3, 3),
plot.chr = "2",
num.plot = 9,
verbose = FALSE)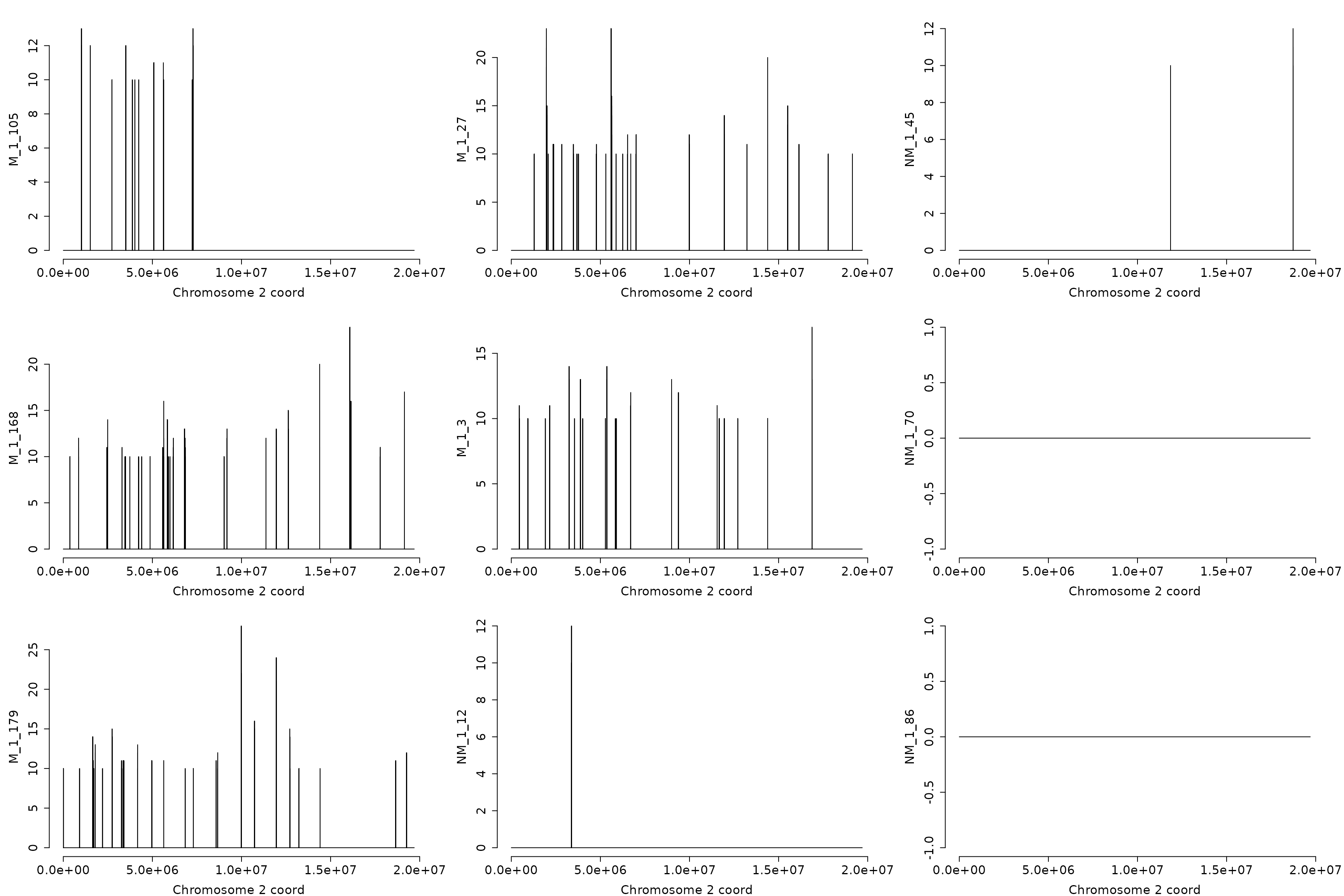
Observe that the same DMR can be present both groups. control and treatment, where some DMPs can share and some not, or simply the DMR can be mostly hypo-methylated in control group and mostly hyper-methylated in the treatment group.
It is the user responsibility to evaluate whether a DMR is specific for one of groups. That is, a further downstream analysis must identify the DMRs that carry discriminatory power to distinguish control individuals from the treatment group, which can be done, e.g., applying a machine learning approach.
References.
Session info
Here is the output of sessionInfo() on the system on
which this document was compiled running pandoc 3.1.1:
## R version 4.3.2 (2023-10-31)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 22.04.3 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.10.0
## LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.10.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: America/New_York
## tzcode source: system (glibc)
##
## attached base packages:
## [1] grid stats4 stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] gridExtra_2.3 ggplot2_3.4.4 MethylIT.utils_0.1.1
## [4] MethylIT_0.3.2.7 rtracklayer_1.60.1 GenomicRanges_1.52.1
## [7] GenomeInfoDb_1.36.4 IRanges_2.34.1 S4Vectors_0.38.2
## [10] BiocGenerics_0.46.0 knitr_1.45
##
## loaded via a namespace (and not attached):
## [1] rstudioapi_0.15.0 jsonlite_1.8.7
## [3] magrittr_2.0.3 modeltools_0.2-23
## [5] rmarkdown_2.25 fs_1.6.3
## [7] BiocIO_1.10.0 zlibbioc_1.46.0
## [9] ragg_1.2.6 vctrs_0.6.5
## [11] memoise_2.0.1 Rsamtools_2.16.0
## [13] RCurl_1.98-1.13 htmltools_0.5.7
## [15] S4Arrays_1.0.6 Formula_1.2-5
## [17] pROC_1.18.5 caret_6.0-94
## [19] sass_0.4.8 parallelly_1.36.0
## [21] bslib_0.6.1 desc_1.4.3
## [23] sandwich_3.0-2 plyr_1.8.9
## [25] zoo_1.8-12 lubridate_1.9.3
## [27] cachem_1.0.8 GenomicAlignments_1.36.0
## [29] lifecycle_1.0.4 minpack.lm_1.2-4
## [31] iterators_1.0.14 pkgconfig_2.0.3
## [33] Matrix_1.6-4 R6_2.5.1
## [35] fastmap_1.1.1 GenomeInfoDbData_1.2.10
## [37] MatrixGenerics_1.12.3 future_1.33.0
## [39] digest_0.6.33 colorspace_2.1-0
## [41] AnnotationDbi_1.62.2 textshaping_0.3.7
## [43] RSQLite_2.3.3 randomForest_4.7-1.1
## [45] fansi_1.0.6 timechange_0.2.0
## [47] httr_1.4.7 abind_1.4-5
## [49] compiler_4.3.2 bit64_4.0.5
## [51] withr_2.5.2 BiocParallel_1.34.2
## [53] betareg_3.1-4 DBI_1.1.3
## [55] highr_0.10 MASS_7.3-60
## [57] lava_1.7.3 DelayedArray_0.26.7
## [59] rjson_0.2.21 ModelMetrics_1.2.2.2
## [61] tools_4.3.2 lmtest_0.9-40
## [63] future.apply_1.11.0 nnet_7.3-19
## [65] glue_1.6.2 restfulr_0.0.15
## [67] nlme_3.1-164 reshape2_1.4.4
## [69] generics_0.1.3 recipes_1.0.8
## [71] gtable_0.3.4 class_7.3-22
## [73] data.table_1.14.8 flexmix_2.3-19
## [75] utf8_1.2.4 XVector_0.40.0
## [77] foreach_1.5.2 pillar_1.9.0
## [79] stringr_1.5.1 genefilter_1.82.1
## [81] splines_4.3.2 dplyr_1.1.4
## [83] lattice_0.22-5 survival_3.5-7
## [85] bit_4.0.5 annotate_1.78.0
## [87] tidyselect_1.2.0 Biostrings_2.68.1
## [89] SummarizedExperiment_1.30.2 xfun_0.41
## [91] Biobase_2.60.0 hardhat_1.3.0
## [93] timeDate_4022.108 matrixStats_1.2.0
## [95] proto_1.0.0 stringi_1.8.3
## [97] yaml_2.3.8 evaluate_0.23
## [99] codetools_0.2-19 tibble_3.2.1
## [101] cli_3.6.2 rpart_4.1.23
## [103] nls2_0.3-3 xtable_1.8-4
## [105] systemfonts_1.0.5 munsell_0.5.0
## [107] jquerylib_0.1.4 Rcpp_1.0.11
## [109] globals_0.16.2 png_0.1-8
## [111] XML_3.99-0.16 parallel_4.3.2
## [113] pkgdown_2.0.7 gower_1.0.1
## [115] blob_1.2.4 bitops_1.0-7
## [117] listenv_0.9.0 ipred_0.9-14
## [119] scales_1.3.0 prodlim_2023.08.28
## [121] purrr_1.0.2 crayon_1.5.2
## [123] rlang_1.1.2 KEGGREST_1.40.1