A function addressed to find DMRs, assuming that differentially methylated position (DMPs) are already identified and carried in 'obj'.
Following MethylIT pipeline, DMRs can be identified in control and in treatment groups. The concept of DMR is subjective in some extend. That is, while MethylIT pipeline identify cytosine positions with high probability to be differential methylated in the treatment group with respect to the control group, it will correspond to the user to define whether to say, e.g., a region 100 bp long carrying 3 DMPs is a DMR. There is not theoretical ground to decide whether such a DMR would be biologically relevant or not for a given species. Only the experimentation or further downstream analyses including, for example, the network enrichment analysis of DMR-associated genes would gives us an answer.
dmrfinder(obj, ...)
# S3 method for list
dmrfinder(
obj,
hypo = FALSE,
hyper = FALSE,
min.signal = 2L,
win.size = 1,
step.size = 1,
grfeatures = NULL,
stat = c("count", "denCount", "density", "mean", "sum", "gmean", "median"),
column = NULL,
tv.colum = 8L,
absolute = FALSE,
select.strand = NULL,
maxgap = -1L,
minoverlap = 0L,
select = "all",
ignore.strand = TRUE,
type = c("within", "start", "end", "equal", "any"),
scaling = 1000L,
logbase = 2,
missings = 0,
naming = FALSE,
na.rm = TRUE,
num.cores = 1L,
tasks = 0,
plots = FALSE,
plot.chr = NULL,
num.plot = length(obj),
xlab = NULL,
ylab = NULL,
mfcol = NULL,
cex.axis = 1.1,
cex.lab = 1.1,
mar = c(4, 5, 2, 2),
verbose = TRUE,
...
)
# S3 method for pDMP
dmrfinder(
obj,
hypo = FALSE,
hyper = FALSE,
min.signal = 2L,
win.size = 1,
step.size = 1,
grfeatures = NULL,
stat = c("sum", "mean", "gmean", "median", "density", "count", "denCount"),
column = NULL,
tv.colum = 8L,
absolute = FALSE,
select.strand = NULL,
maxgap = -1L,
minoverlap = 0L,
select = "all",
ignore.strand = TRUE,
type = c("within", "start", "end", "equal", "any"),
scaling = 1000L,
logbase = 2,
missings = 0,
naming = FALSE,
na.rm = TRUE,
num.cores = 1L,
tasks = 0,
plots = FALSE,
plot.chr = NULL,
num.plot = length(obj),
xlab = NULL,
ylab = NULL,
mfcol = NULL,
cex.axis = 1.5,
cex.lab = 1.5,
mar = c(4, 5, 2, 2),
verbose = TRUE,
...
)
# S3 method for InfDiv
dmrfinder(
obj,
hypo = FALSE,
hyper = FALSE,
min.signal = 2L,
win.size = 1,
step.size = 1,
grfeatures = NULL,
stat = c("sum", "mean", "gmean", "median", "density", "count", "denCount"),
column = NULL,
tv.colum = 8L,
absolute = FALSE,
select.strand = NULL,
maxgap = -1L,
minoverlap = 0L,
select = "all",
ignore.strand = TRUE,
type = c("within", "start", "end", "equal", "any"),
scaling = 1000L,
logbase = 2,
missings = 0,
naming = FALSE,
na.rm = TRUE,
num.cores = 1L,
tasks = 0,
plots = FALSE,
plot.chr = NULL,
num.plot = length(obj),
xlab = NULL,
ylab = NULL,
mfcol = NULL,
cex.axis = 1.5,
cex.lab = 1.5,
mar = c(4, 5, 2, 2),
verbose = TRUE,
...
)Arguments
- obj
An object from pDMP or InfDiv class or simply a list of GRanges-class.
- ...
If plots == TRUE, then further parameter for plot can be passed.
- hypo
Logical. If TRUE then only hypo-methylated positions are considered.
- hyper
Logical. If TRUE then only hyper-methylated positions are considered.
- min.signal
A number denoting minimun number of differentially methylated positions (DMPs) or signal value (if stat != "count") that must be in region to be considered as a potential differentially methylated region (DMR).
- win.size, step.size, grfeatures, stat, column
Described in getGRangesStat.
- tv.colum
Only if hypo or hyper is TRUE. An integer denoting whether the column carrying the difference of methylation levels (total variance, TV) is locate on each
GRanges-classobject from 'obj'.- absolute, select.strand, maxgap, minoverlap
Described in getGRangesStat.
- select, ignore.strand, type, scaling, logbase
Described in getGRangesStat.
- missings, naming, na.rm, num.cores, tasks
Described in getGRangesStat.
- plots
Logical. Whether to plot a graphic showing the identified DMRs (if any) on a specified chromosome coordinates.
- plot.chr
DMRs found on different chromosomes would have the same numerical coordinate and, consequently to show the graphics, we must specify the chromosome.
- num.plot
If plots = TRUE, an integer denoting the number of plots.
- xlab, ylab, mfcol, cex.lab, mar, verbose
Described in getGRangesStat.
Value
A GRanges carrying all the identified DMRs. The indexes of the regions holding the condition sets by 'min.signal' can be retrieved by typing 'metadata(x)'.
Details
There is not a unique setting to find DMRs. Under different settings clusterization algorithm can yield different set of DMRs. The users are responsible to find which are the best setting for them. A DMR found in the treatment group does not implies that the same DMR is not in the control group. Indeed, for example, a DMR found in the treatment group would be also a DMR in the control group, where some DMPs can share and some not, or simply the DMR can be mostly hypo-methylated in control group and mostly hyper-methylated in the treatment group.
Examples
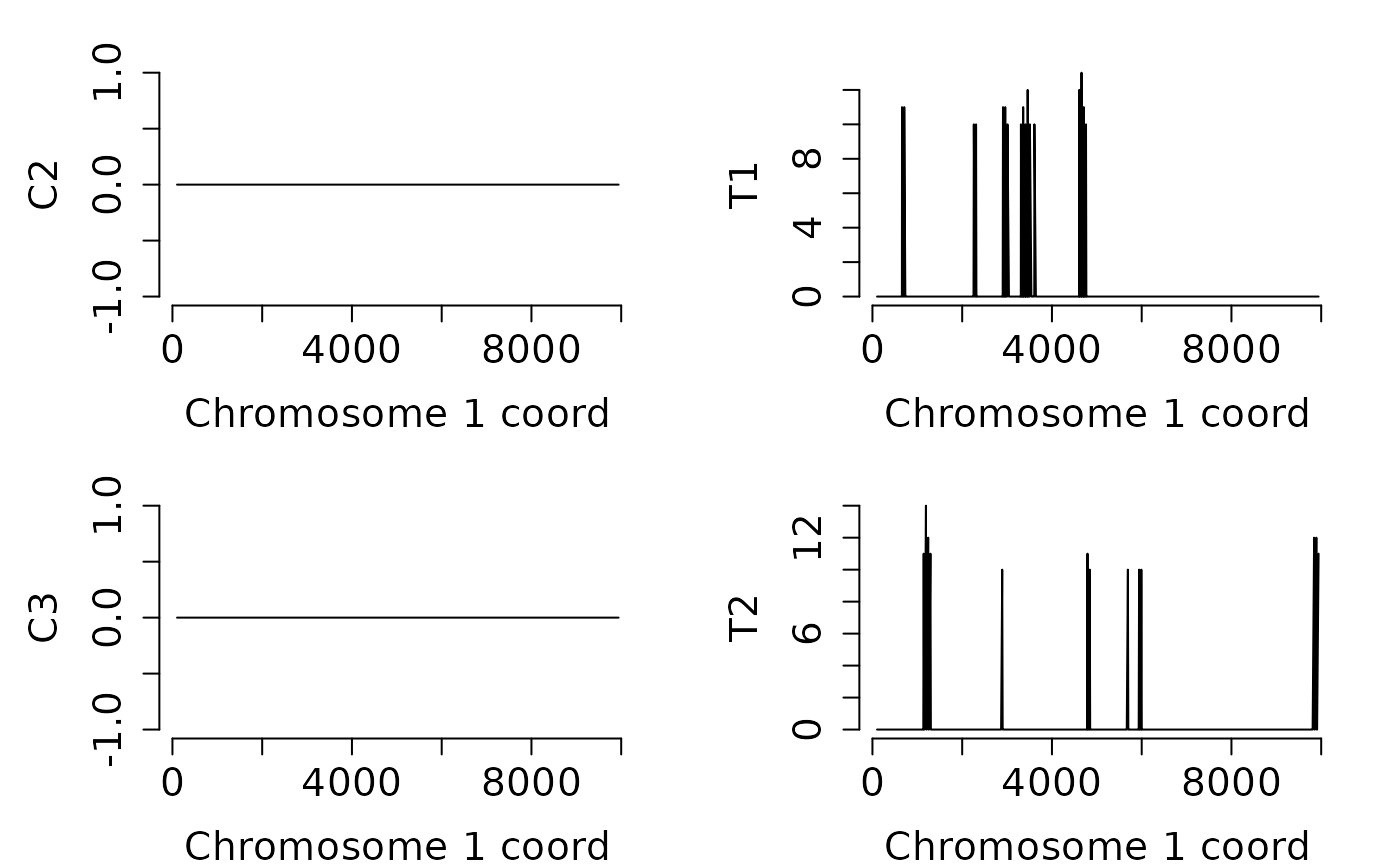
## Load a DMPs example data set
data(dmps, package = "MethylIT")
## Search hypo-methylated DMRs with at least two DMPs on 200 bp.
## The graphics of samples 2 to 5 are returned and
## they indicate that there is not hypo-DMRs with such
## features in neither of the groups, control and treatment.
hyp_dmrs <- dmrfinder(
obj = dmps,
hypo = TRUE,
min.signal = 2L,
win.size = 200,
step.size = 50,
stat = "count",
tv.colum = 8L,
naming = TRUE,
plots = TRUE,
mfcol = c(2, 2),
num.plot = c(2:5),
verbose = FALSE)
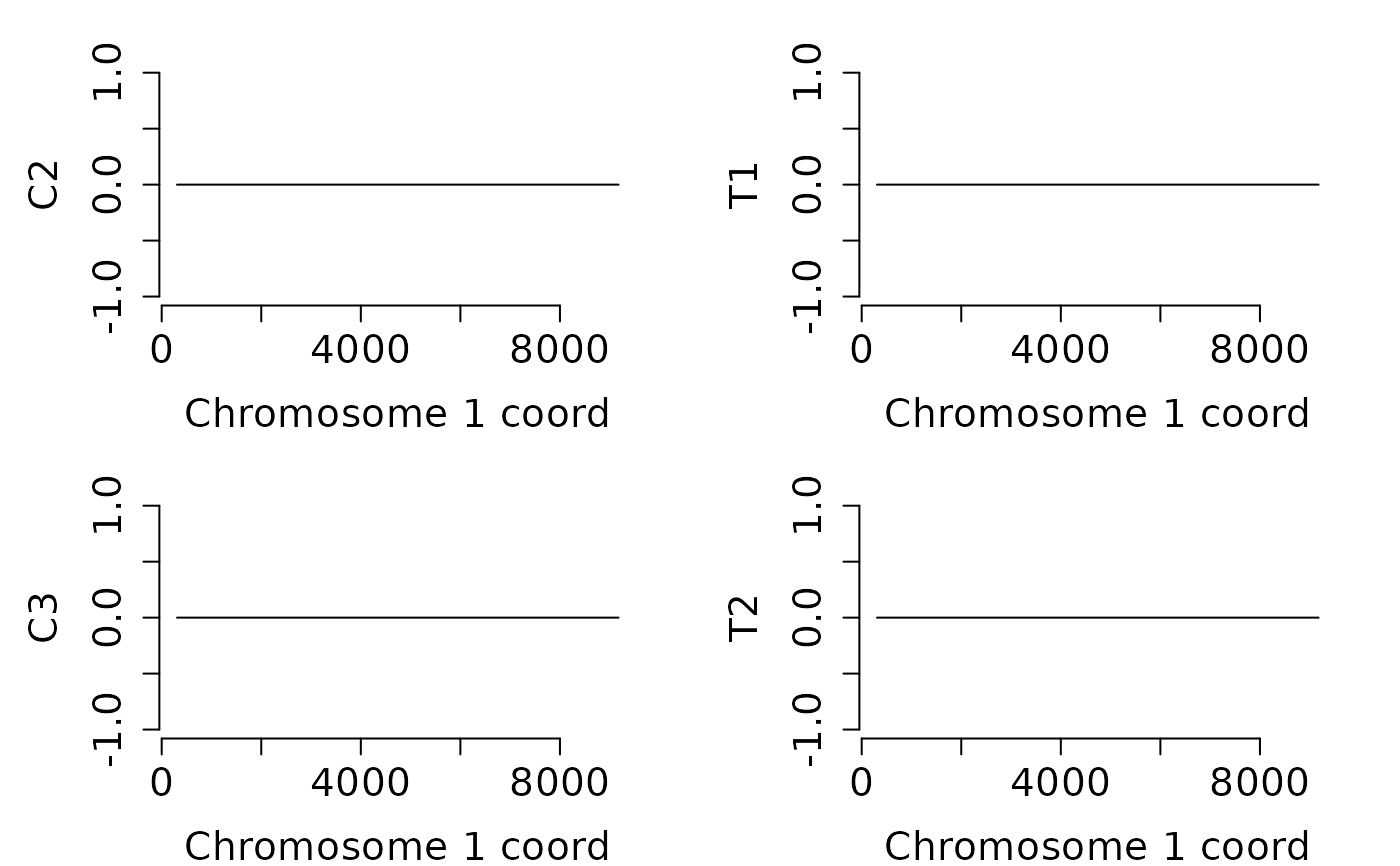 ## Search hyper-methylated DMRs with at least two DMPs on 200 bp.
## Only the treatment group carries hyper-DMRs with such features.
hyp_dmrs <- dmrfinder(
obj = dmps,
hyper = TRUE,
min.signal = 10L,
win.size = 200,
step.size = 50,
stat = "count",
tv.colum = 8L,
naming = TRUE,
plots = TRUE,
mfcol = c(2, 2),
num.plot = c(2:5),
verbose = FALSE)
## Search hyper-methylated DMRs with at least two DMPs on 200 bp.
## Only the treatment group carries hyper-DMRs with such features.
hyp_dmrs <- dmrfinder(
obj = dmps,
hyper = TRUE,
min.signal = 10L,
win.size = 200,
step.size = 50,
stat = "count",
tv.colum = 8L,
naming = TRUE,
plots = TRUE,
mfcol = c(2, 2),
num.plot = c(2:5),
verbose = FALSE)