A Short Introduction to Algebraic Taxonomy on Genes Regions
Robersy Sanchez
Department of Biology. Pennsylvania State University, University Park, PA 16802rus547@psu.edu
28 December 2024
Source:vignettes/automorphism_and_decision_tree.Rmd
automorphism_and_decision_tree.RmdAbstract
Herein, we show a short and simply introduction to algebraic taxonomy of genomic/genes regions based on the nalysis of DNA mutational events on DNA Multiple Sequence Alignment (MSA) by means of automorphisms between pairwise DNA sequences algebraically represented as Abelian finite group.
Overview This is a R package to compute the autimorphisms between pairwise
aligned DNA sequences represented as elements from a Genomic Abelian group as described in reference (1). In a general scenario, whole chromosomes or genomic regions from a population (from any species or close related species) can be algebraically represented as a direct sum of cyclic groups or more specifically Abelian p-groups. Basically, we propose the representation of multiple sequence alignments (MSA) of length N as a finite Abelian group created by the direct sum of Abelian group of prime-power order:
Where, the ’s are prime numbers, and is the group of integer modulo .
For the purpose of estimating the automorphism between two aligned DNA sequences, .
Automorphisms
Herein, automorphisms are considered algebraic descriptions of mutational event observed in codon sequences represented on different Abelian groups. In particular, as described in references (3-4), for each representation of the codon set on a defined Abelian group there are 24 possible isomorphic Abelian groups. These Abelian groups can be labeled based on the DNA base-order used to generate them. The set of 24 Abelian groups can be described as a group isomorphic to the symmetric group of degree four (, see reference (4)).
For further support about the symmetric group on the 24 Abelian group of genetic-code cubes, users can also see Symmetric Group of the Genetic-Code Cubes., specifically the Mathematica notebook IntroductionToZ5GeneticCodeVectorSpace.nb and interact with it using Wolfram Player, freely available (for Windows and Linux OS) at, https://www.wolfram.com/player/.
Automorphisms on
First, we proceed to load or install, if needed, the R packages required for our analysis. If needed the R package required for the current analysis can be installed as:
install.packages(c("party", "partykit", "data.table", "ggplot2",
"ggparty", "dplyr"), dependencies=TRUE)In particular, you might require to install the library CHAID can be installed typing in R console:
install.packages("CHAID", repos="http://R-Forge.R-project.org")You can install GenomAutomorphism package from GitHub
devtools::install_git("https://github.com/genomaths/GenomAutomorphism.git")If all the required libraries all installed, then we proceed to load the libraries
library(GenomAutomorphism)
library(Biostrings)
library(party)
library(partykit)
library(data.table)
library(ggplot2)
library(ggparty)
library(dplyr)
library(CHAID)Next, we proceed to check the DNA multiple sequence alignment (MSA) file. This is a FASTA file carrying the MSA of primate BRCA1 DNA repair gene. Notice that we are familiar with the FASTA file, then it is better to directly read it with function automorphisms. However, for the current example, this step can be bypassed, since the MSA is provided provided together with GenomAutomorphism R package
## Do not run it. This is included with package
URL <- paste0("https://github.com/genomaths/seqalignments/raw/master/BRCA1/",
"brca1_primates_dna_repair_20_sequences.fasta")
brca1_aln <- readDNAMultipleAlignment(filepath = URL)Load MSA available in the package
data("brca1_aln", package = "GenomAutomorphism")
brca1_aln
#> DNAMultipleAlignment with 20 rows and 2283 columns
#> aln names
#> [1] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA NM_007298.3:20-22...
#> [2] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA U64805.1:1-2280_H...
#> [3] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA XM_031011560.1:23...
#> [4] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA XM_031011561.1:23...
#> [5] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA XM_031011562.1:16...
#> [6] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCTCAGATCCCCCACAGCCACTACTGA XM_009432101.3:27...
#> [7] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCTCAGATCCCCCACAGCCACTACTGA XM_009432104.3:37...
#> [8] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCTCAGATCCCCCACAGCCACTACTGA XM_016930487.2:37...
#> [9] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCTCAGATCCCCCACAGCCACTACTGA XM_009432099.3:37...
#> ... ...
#> [12] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCTCAGATCCCCCACAGCCACTACTGA XM_034941185.1:24...
#> [13] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCTCAGATCCCCCACAGCCACTACTGA XM_034941182.1:25...
#> [14] ATGGATTTATCTGCTGTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGATACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA XM_032163757.1:14...
#> [15] ATGGATTTATCTGCTGTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGATACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA XM_032163756.1:14...
#> [16] ATGGATTTATCTGCTGTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGATACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA XM_032163758.1:13...
#> [17] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACGCCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA XM_030923119.1:18...
#> [18] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACGCCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA XM_030923118.1:18...
#> [19] ATGGATTTACCTGCTGTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA XM_025363316.1:14...
#> [20] ATGGATTTATCTGCTGTTCGTGTTGAAGAAGTGCAAAATGTCCTT...CTGGACACCTACCTGATACCCCAGATCCCTCACAGCCACTACTGA XM_039475995.1:49...The sequence names
strtrim(names(brca1_aln@unmasked), 100)
#> [1] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_DNA_repair_associated_(BRCA1)_transcript_variant_4_mRNA"
#> [2] "U64805.1:1-2280_Homo_sapiens_Brca1-delta11b_(Brca1)_mRNA_complete_cds"
#> [3] "XM_031011560.1:233-2515_PREDICTED:_Gorilla_gorilla_gorilla_BRCA1_DNA_repair_associated_(BRCA1)_trans"
#> [4] "XM_031011561.1:233-2512_PREDICTED:_Gorilla_gorilla_gorilla_BRCA1_DNA_repair_associated_(BRCA1)_trans"
#> [5] "XM_031011562.1:163-2442_PREDICTED:_Gorilla_gorilla_gorilla_BRCA1_DNA_repair_associated_(BRCA1)_trans"
#> [6] "XM_009432101.3:276-2555_PREDICTED:_Pan_troglodytes_BRCA1_DNA_repair_associated_(BRCA1)_transcript_va"
#> [7] "XM_009432104.3:371-2650_PREDICTED:_Pan_troglodytes_BRCA1_DNA_repair_associated_(BRCA1)_transcript_va"
#> [8] "XM_016930487.2:371-2650_PREDICTED:_Pan_troglodytes_BRCA1_DNA_repair_associated_(BRCA1)_transcript_va"
#> [9] "XM_009432099.3:371-2653_PREDICTED:_Pan_troglodytes_BRCA1_DNA_repair_associated_(BRCA1)_transcript_va"
#> [10] "XM_034941183.1:254-2533_PREDICTED:_Pan_paniscus_BRCA1_DNA_repair_associated_(BRCA1)_transcript_varia"
#> [11] "XM_034941184.1:254-2533_PREDICTED:_Pan_paniscus_BRCA1_DNA_repair_associated_(BRCA1)_transcript_varia"
#> [12] "XM_034941185.1:248-2527_PREDICTED:_Pan_paniscus_BRCA1_DNA_repair_associated_(BRCA1)_transcript_varia"
#> [13] "XM_034941182.1:254-2536_PREDICTED:_Pan_paniscus_BRCA1_DNA_repair_associated_(BRCA1)_transcript_varia"
#> [14] "XM_032163757.1:145-2418_PREDICTED:_Hylobates_moloch_BRCA1_DNA_repair_associated_(BRCA1)_transcript_v"
#> [15] "XM_032163756.1:145-2421_PREDICTED:_Hylobates_moloch_BRCA1_DNA_repair_associated_(BRCA1)_transcript_v"
#> [16] "XM_032163758.1:139-2412_PREDICTED:_Hylobates_moloch_BRCA1_DNA_repair_associated_(BRCA1)_transcript_v"
#> [17] "XM_030923119.1:184-2463_PREDICTED:_Rhinopithecus_roxellana_BRCA1_DNA_repair_associated_(BRCA1)_trans"
#> [18] "XM_030923118.1:183-2465_PREDICTED:_Rhinopithecus_roxellana_BRCA1_DNA_repair_associated_(BRCA1)_trans"
#> [19] "XM_025363316.1:147-2426_PREDICTED:_Theropithecus_gelada_BRCA1_DNA_repair_associated_(BRCA1)_transcri"
#> [20] "XM_039475995.1:49-2328_PREDICTED:_Saimiri_boliviensis_boliviensis_BRCA1_DNA_repair_associated_(BRCA1"Next, function automorphisms will be applied to represent the codon sequence in the Abelian group (i.e., the set of integers remainder modulo 64). The codon coordinates are requested on the cube ACGT. Following reference (4)), cubes are labeled based on the order of DNA bases used to define the sum operation.
In Z64, automorphisms are described as functions , where and are elements from the set of integers modulo 64. Below, in function automorphisms three important arguments are given values: group = “Z64”, cube = c(“ACGT”, “TGCA”), and cube_alt = c(“CATG”, “GTAC”). Setting for group specifies on which group the automorphisms will be computed. These groups can be: “Z5”, “Z64”, “Z125”, and “Z5^3”.
In groups “Z64” and “Z125” not all the mutational events can be described as automorphisms from a given cube. So, a character string denoting pairs of “dual” the genetic-code cubes, as given in references (1-4)), is given as argument for cube. That is, the base pairs from the given cubes must be complementary each other. Such a cube pair are call dual cubes and, as shown in reference (4)), each pair integrates group. If automorphisms are not found in first set of dual cubes, then the algorithm search for automorphisms in a alternative set of dual cubes.
## Do not run it. This is included with package
nams <- c("human_1","human_2","gorilla_1","gorilla_2","gorilla_3",
"chimpanzee_1","chimpanzee_2","chimpanzee_3","chimpanzee_4",
"bonobos_1","bonobos_2","bonobos_3","bonobos_4","silvery_gibbon_1",
"silvery_gibbon_1","silvery_gibbon_3","golden_monkey_1",
"golden_monkey_2","gelada_baboon","bolivian_monkey")
brca1_autm <- automorphisms(
seqs = brca1_aln,
group = "Z64",
cube = c("ACGT", "TGCA"),
cube_alt = c("CATG", "GTAC"),
nms = nams,
verbose = FALSE)Object brca1_autm is included with package and can be load typing:
data(brca1_autm, package = "GenomAutomorphism")
brca1_autm
#> AutomorphismList object of length: 190
#> names(190): human_1.human_2 human_1.gorilla_1 human_1.gorilla_2 ... golden_monkey_2.gelada_baboon golden_monkey_2.bolivian_monkey gelada_baboon.bolivian_monkey
#> -------
#> Automorphism object with 761 ranges and 8 metadata columns:
#> seqnames ranges strand | seq1 seq2 aa1 aa2 coord1 coord2 autm
#> <Rle> <IRanges> <Rle> | <character> <character> <character> <character> <numeric> <numeric> <numeric>
#> [1] 1 1 + | ATG ATG M M 50 50 1
#> [2] 1 2 + | GAT GAT D D 11 11 1
#> [3] 1 3 + | TTA TTA L L 60 60 1
#> [4] 1 4 + | TCT TCT S S 31 31 1
#> [5] 1 5 + | GCT GCT A A 27 27 1
#> ... ... ... ... . ... ... ... ... ... ... ...
#> [757] 1 757 + | CAC CAC H H 5 5 1
#> [758] 1 758 + | AGC AGC S S 33 33 1
#> [759] 1 759 + | CAC CAC H H 5 5 1
#> [760] 1 760 + | TAC TAC Y Y 13 13 1
#> [761] 1 761 + | TGA TGA * * 44 44 1
#> cube
#> <character>
#> [1] ACGT
#> [2] ACGT
#> [3] ACGT
#> [4] ACGT
#> [5] ACGT
#> ... ...
#> [757] ACGT
#> [758] ACGT
#> [759] ACGT
#> [760] ACGT
#> [761] ACGT
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths
#> ...
#> <189 more DFrame element(s)>
#> Two slots: 'DataList' & 'SeqRanges'
#> -------Grouping automorphisms by automorphism’s coefficients
Automorphisms with the same automorphism’s coefficients can be grouped. This task can be accomplished with function automorphism_bycoef. However, for the sake of time, its output is included in the package
## Not need to run it here
autby_coef <- automorphism_bycoef(x = brca1_autm,
verbose = FALSE)Object brca1_autm is included with package and can be load typing:
data(autby_coef, package = "GenomAutomorphism")
autby_coef
#> AutomorphismByCoefList object of length 190:
#> $human_1.human_2
#> AutomorphismByCoef object with 239 ranges and 7 metadata columns:
#> seqnames ranges strand | seq1 seq2 aa1 aa2 autm mut_type cube
#> <Rle> <IRanges> <Rle> | <character> <character> <character> <character> <numeric> <character> <character>
#> [1] 1 1-238 + | ATG ATG M M 1 HHH ACGT
#> [2] 1 1-238 + | GAT GAT D D 1 HHH ACGT
#> [3] 1 1-238 + | TTA TTA L L 1 HHH ACGT
#> [4] 1 1-238 + | TCT TCT S S 1 HHH ACGT
#> [5] 1 1-238 + | GCT GCT A A 1 HHH ACGT
#> ... ... ... ... . ... ... ... ... ... ... ...
#> [235] 1 511-761 + | CCC CCC P P 1 HHH ACGT
#> [236] 1 511-761 + | CTT CTT L L 1 HHH ACGT
#> [237] 1 511-761 + | CCT CCT P P 1 HHH ACGT
#> [238] 1 511-761 + | ATA ATA I I 1 HHH ACGT
#> [239] 1 511-761 + | TGA TGA * * 1 HHH ACGT
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths
#>
#> ...
#> <189 more elements>In the next we are interested on mutational events in respect to human (as reference).
nams <- names(brca1_autm)
idx1 <- grep("human_1.", nams)
idx2 <- grep("human_2.", nams)
idx <- union(idx1, idx2)
h_brca1_autm <- unlist(brca1_autm[ idx ])
h_brca1_autm = h_brca1_autm[ which(h_brca1_autm$autm != 1) ]
h_brca1_autm
#> Automorphism object with 1397 ranges and 8 metadata columns:
#> seqnames ranges strand | seq1 seq2 aa1 aa2 coord1
#> <Rle> <IRanges> <Rle> | <character> <character> <character> <character> <numeric>
#> human_1.human_2 1 239 + | CAT CGT H R 7
#> human_1.human_2 1 253 + | GCA GTA A V 24
#> human_1.human_2 1 323 + | TCT CCT S P 31
#> human_1.human_2 1 333 + | TCT TCC S S 31
#> human_1.human_2 1 350 + | --- --- - - NA
#> ... ... ... ... . ... ... ... ... ...
#> human_2.bolivian_monkey 1 716 + | AAT AGT N S 3
#> human_2.bolivian_monkey 1 726 + | GAG GAA E E 10
#> human_2.bolivian_monkey 1 730 + | GTG GTA V V 58
#> human_2.bolivian_monkey 1 731 + | ACC ACT T T 17
#> human_2.bolivian_monkey 1 756 + | CCC CCT P P 21
#> coord2 autm cube
#> <numeric> <numeric> <character>
#> human_1.human_2 39 33 ACGT
#> human_1.human_2 56 21 ACGT
#> human_1.human_2 23 9 ACGT
#> human_1.human_2 29 3 ACGT
#> human_1.human_2 NA -1 Gaps
#> ... ... ... ...
#> human_2.bolivian_monkey 35 33 ACGT
#> human_2.bolivian_monkey 8 52 ACGT
#> human_2.bolivian_monkey 56 12 ACGT
#> human_2.bolivian_monkey 19 35 ACGT
#> human_2.bolivian_monkey 23 59 ACGT
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengthsBar plot automorphism distribution by coefficient
The automorphism distribution by cubes can be summarized in the bar-plot graphic.
Object autby_coef carried all the pairwise comparisons, while it will be enough to use data from a single species as reference, e.g., humans.
First the data must be reordered into a object:
h_autby_coef <- automorphism_bycoef(x = h_brca1_autm)
h_autby_coef
#> AutomorphismByCoef object with 1395 ranges and 7 metadata columns:
#> seqnames ranges strand | seq1 seq2 aa1 aa2 autm
#> <Rle> <IRanges> <Rle> | <character> <character> <character> <character> <numeric>
#> human_1.gelada_baboon 1 4 + | TCT CCT S P 9
#> human_2.gelada_baboon 1 4 + | TCT CCT S P 9
#> human_1.silvery_gibbon_1 1 6 + | CTT GTT L V 29
#> human_1.silvery_gibbon_1 1 6 + | CTT GTT L V 29
#> human_1.silvery_gibbon_3 1 6 + | CTT GTT L V 29
#> ... ... ... ... . ... ... ... ... ...
#> human_2.bonobos_2 1 753 + | CCC CCT P P 59
#> human_2.bonobos_3 1 753 + | CCC CCT P P 59
#> human_2.bonobos_4 1 753 + | CCC CCT P P 59
#> human_1.bolivian_monkey 1 756 + | CCC CCT P P 59
#> human_2.bolivian_monkey 1 756 + | CCC CCT P P 59
#> mut_type cube
#> <character> <character>
#> human_1.gelada_baboon YHH ACGT
#> human_2.gelada_baboon YHH ACGT
#> human_1.silvery_gibbon_1 SHH ACGT
#> human_1.silvery_gibbon_1 SHH ACGT
#> human_1.silvery_gibbon_3 SHH ACGT
#> ... ... ...
#> human_2.bonobos_2 HHY ACGT
#> human_2.bonobos_3 HHY ACGT
#> human_2.bonobos_4 HHY ACGT
#> human_1.bolivian_monkey HHY ACGT
#> human_2.bolivian_monkey HHY ACGT
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengthsEvery single base mutational event across the MSA was classified according IUPAC nomenclature: 1) According to the number of hydrogen bonds (on DNA/RNA double helix): strong S={C, G} (three hydrogen bonds) and weak W={A, U} (two hydrogen bonds). According to the chemical type: purines R={A, G} and pyrimidines Y={C, U}. 3). According to the presence of amino or keto groups on the base rings: amino M={C, A} and keto K={G, T}. Constant (hold) base positions were labeled with letter H. So, codon positions labeled as HKH means that the first and third bases remains constant and mutational events between bases G and T were found in the MSA.
nams <- names(h_autby_coef)
nams <- sub("human[_][1-2][.]", "", nams)
nams <- sub("[_][1-6]", "", nams)
dt <- data.frame(h_autby_coef, species = nams)
dt <- data.frame(dt, species = nams)
dt <- dt[, c("start", "autm", "species", "mut_type", "cube")]
DataFrame(dt)
#> DataFrame with 1395 rows and 5 columns
#> start autm species mut_type cube
#> <integer> <numeric> <character> <character> <character>
#> 1 4 9 gelada_baboon YHH ACGT
#> 2 4 9 gelada_baboon YHH ACGT
#> 3 6 29 silvery_gibbon SHH ACGT
#> 4 6 29 silvery_gibbon SHH ACGT
#> 5 6 29 silvery_gibbon SHH ACGT
#> ... ... ... ... ... ...
#> 1391 753 59 bonobos HHY ACGT
#> 1392 753 59 bonobos HHY ACGT
#> 1393 753 59 bonobos HHY ACGT
#> 1394 756 59 bolivian_monkey HHY ACGT
#> 1395 756 59 bolivian_monkey HHY ACGTNominal variables are transformed into
dt$start <- as.numeric(dt$start)
dt$autm <- as.numeric(dt$autm)
dt$cube <- as.factor(dt$cube)
dt$species <- as.factor(dt$species)
dt$mut_type <- as.factor(dt$mut_type)
counts <- table(dt$cube)
par(family = "serif", cex = 0.6, font = 2, mar=c(4,6,4,4))
barplot(counts, #main="Automorphism distribution",
xlab="Genetic-code cube representation",
ylab="Fixed mutational events",
col=c("darkblue","red", "darkgreen", "magenta", "orange"),
border = NA, axes = F, #ylim = c(0, 6000),
cex.lab = 2, cex.main = 1.5, cex.names = 2)
axis(2, at = c(0, 200, 400, 600, 800, 1000), cex.axis = 1.5)
mtext(side = 1,line = -2.8, at = c(0.7, 1.9, 3.1, 4.3),
text = paste0( counts ), cex = 1.4,
col = c("white", "red","yellow", "black"))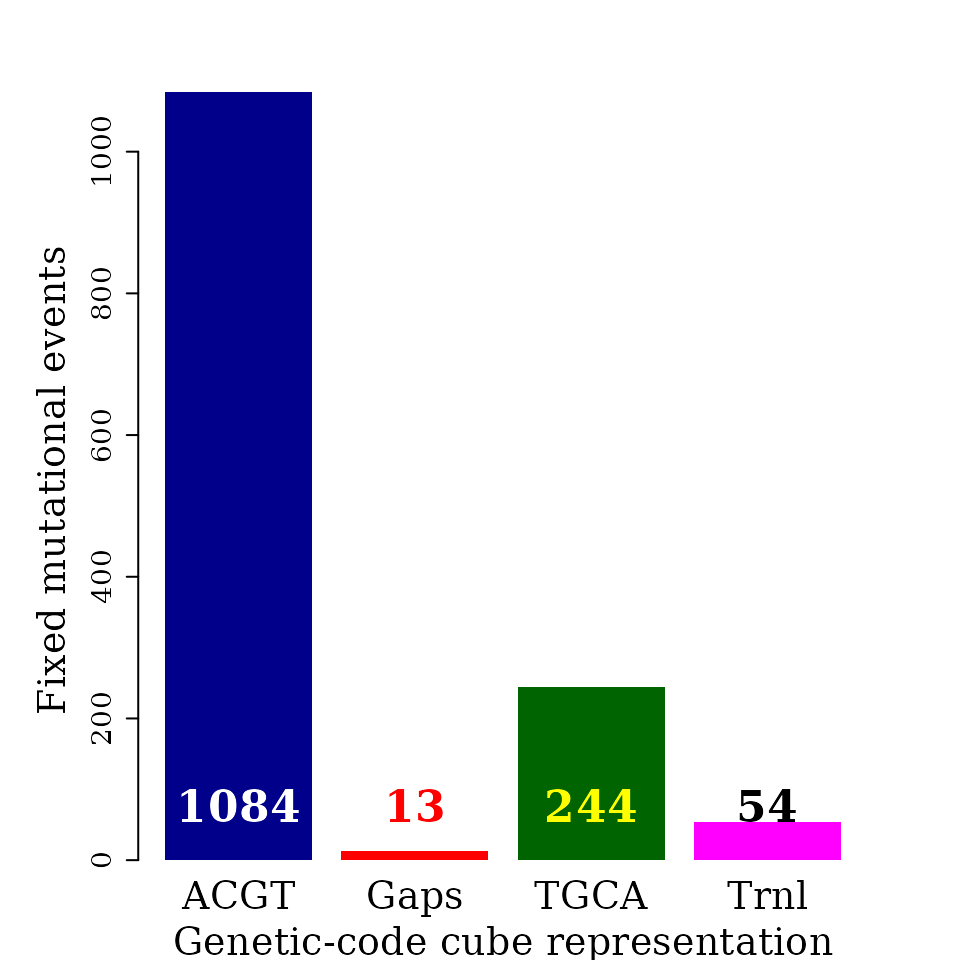
Classification Tree Chi-squared Automated Interaction Detection (CHAID)
The current CHAID implementation only accepts nominal or ordinal categorical predictors. When predictors are continuous, they have to be transformed into ordinal predictors before using the following algorithm. We create a ordinal variable autms from variable autm. CHAID can be download at: <install.packages(“CHAID”, repos=“http://R-Forge.R-project.org”)>
interval <- function(x, a, b) {
x >= a & x <= b
}
datos = dt
datos$autms <- case_when(datos$autm < 16 ~ 'A1',
interval(datos$autm, 16, 31) ~ 'A2',
interval(datos$autm, 32, 47) ~ 'A3',
datos$autm > 47 ~ 'A4')
datos$autms <- as.factor(datos$autms)
datos$mut_type <- as.character(datos$mut_type)
datos$mut_type[ which(datos$cube == "Trnl") ] <- "indel"
datos$mut_type[ which(datos$cube == "Gaps") ] <- "---"
datos$mut_type <- as.factor(datos$mut_type)
datos$regions <- case_when(datos$start < 230 ~ 'R0',
interval(datos$start, 230, 270) ~ 'R1',
interval(datos$start, 271, 305) ~ 'R2',
interval(datos$start, 306, 338) ~ 'R3',
interval(datos$start, 339, 533) ~ 'R4',
interval(datos$start, 534, 570) ~ 'R5',
interval(datos$start, 571, 653) ~ 'R6',
interval(datos$start, 654, 709) ~ 'R7',
datos$start > 709 ~ 'R8')
datos$regions <- as.factor(datos$regions)
datos$autm <- as.factor(datos$autm)
datos$species <- as.factor(datos$species)
datos$start <- as.factor(datos$start)
datos$cube <- as.factor(datos$cube)
datos <- datos[, c( "autms", "regions", "mut_type", "cube", "species")]
DataFrame(datos)
#> DataFrame with 1395 rows and 5 columns
#> autms regions mut_type cube species
#> <factor> <factor> <factor> <factor> <factor>
#> 1 A1 R0 YHH ACGT gelada_baboon
#> 2 A1 R0 YHH ACGT gelada_baboon
#> 3 A2 R0 SHH ACGT silvery_gibbon
#> 4 A2 R0 SHH ACGT silvery_gibbon
#> 5 A2 R0 SHH ACGT silvery_gibbon
#> ... ... ... ... ... ...
#> 1391 A4 R8 HHY ACGT bonobos
#> 1392 A4 R8 HHY ACGT bonobos
#> 1393 A4 R8 HHY ACGT bonobos
#> 1394 A4 R8 HHY ACGT bolivian_monkey
#> 1395 A4 R8 HHY ACGT bolivian_monkeyA classification tree is estimated with CHAID algorithm:
ctrl <- chaid_control(minsplit = 200, minprob = 0.8, alpha2 = 0.01, alpha4 = 0.01)
chaid_res <- chaid(species ~ autms + regions + mut_type + cube , data = datos,
control = ctrl)
chaid_res
#>
#> Model formula:
#> species ~ autms + regions + mut_type + cube
#>
#> Fitted party:
#> [1] root
#> | [2] mut_type in ---, HRH: golden_monkey (n = 169, err = 81.1%)
#> | [3] mut_type in HHK, HHR, HHW, HMR, SHH, YHH
#> | | [4] mut_type in ---, HHK, HHM, HHR, HHS, HHY, HKH, HMH, HMR, HRH, HRR, HRW, HRY, HSH, HWH, HYH, HYS, indel, KHH, MHH, RHH, RMH, WHH, WHK, YRH, YRY
#> | | | [5] regions in R0, R4: bolivian_monkey (n = 142, err = 73.2%)
#> | | | [6] regions in R1: gorilla (n = 20, err = 70.0%)
#> | | | [7] regions in R2, R3, R6: golden_monkey (n = 34, err = 41.2%)
#> | | | [8] regions in R5, R7, R8: bolivian_monkey (n = 24, err = 41.7%)
#> | | [9] mut_type in HHW, SHH, YHH: silvery_gibbon (n = 180, err = 66.7%)
#> | [10] mut_type in HHM, HKH, HWH, HYS, WHH: golden_monkey (n = 72, err = 44.4%)
#> | [11] mut_type in HHS, HHY, HMH, HRR, HRW, HYH, MHH, RHH, YRH
#> | | [12] autms in A1
#> | | | [13] regions in R0: golden_monkey (n = 50, err = 76.0%)
#> | | | [14] regions in R1, R2, R4, R5, R8: golden_monkey (n = 144, err = 58.3%)
#> | | | [15] regions in R3, R7: silvery_gibbon (n = 31, err = 41.9%)
#> | | | [16] regions in R6: gorilla (n = 12, err = 50.0%)
#> | | [17] autms in A2: golden_monkey (n = 67, err = 70.1%)
#> | | [18] autms in A3, A4
#> | | | [19] regions in R0, R1, R3, R4, R8
#> | | | | [20] mut_type in ---, HHK, HHM, HHR, HHS, HHW, HKH, HMH, HMR, HRH, HRR, HRW, HRY, HSH, HWH, HYS, indel, KHH, RHH, RMH, SHH, WHH, WHK, YHH, YRH, YRY: golden_monkey (n = 182, err = 79.1%)
#> | | | | [21] mut_type in HHY, HYH: silvery_gibbon (n = 132, err = 77.3%)
#> | | | | [22] mut_type in MHH: gorilla (n = 8, err = 25.0%)
#> | | | [23] regions in R2, R7: bolivian_monkey (n = 18, err = 33.3%)
#> | | | [24] regions in R5, R6: golden_monkey (n = 14, err = 14.3%)
#> | [25] mut_type in HRY, HSH, KHH, RMH, WHK: bolivian_monkey (n = 40, err = 55.0%)
#> | [26] mut_type in indel, YRY: silvery_gibbon (n = 56, err = 67.9%)
#>
#> Number of inner nodes: 7
#> Number of terminal nodes: 19Plotting the CHAID tree
Next, the data must be prepared for plotting the tree with ggparty:
## Updating CHAID decision tree
dp <- data_party(chaid_res)
dat <- dp[, c("autms", "regions", "mut_type", "cube")]
dat$species <- dp[, "(response)"]
chaid_tree <- party(node = node_party(chaid_res),
data = dat,
fitted = dp[, c("(fitted)", "(response)")],
names = names(chaid_res))
## Extract p-values
pvals <- unlist(nodeapply(chaid_tree, ids = nodeids(chaid_tree), function(n) {
pvals <- info_node(n)$adjpvals
pvals < pvals[ which.min(pvals) ]
return(pvals)
}))
pvals <- pvals[ pvals < 0.05 ]
## Counts of event per spciees on each node
node.freq <- sapply(seq_along(chaid_tree), function(id) {
y <- data_party(chaid_tree, id = id)
y <- y[[ "(response)" ]]
table(y)
})
## total counts on each
node.size = colSums(node.freq)Plotting the tree with ggparty (font size adjusted for html output)
ggparty(chaid_tree) +
geom_edge(aes(color = id, size = node.size[id]/300), show.legend = FALSE) +
geom_edge_label(size = 14, colour = "red",
fontface = "bold",
shift = 0.64,
nudge_x = -0.01,
max_length = 10,
splitlevels = 1:4) +
geom_node_label(line_list = list(aes(label = paste0("Node ", id, ": ", splitvar)),
aes(label = paste0("N=", node.size[id], ", p",
ifelse(pvals < .001, "<.001",
paste0("=", round(pvals, 3)))),
size = 30)),
line_gpar = list(list(size = 30),
list(size = 30)),
ids = "inner", fontface = "bold", size = 30) +
geom_node_info() +
geom_node_label(aes(label = paste0("N = ", node.size),
fontface = "bold"),
ids = "terminal", nudge_y = -0.0, nudge_x = 0.01, size = 12) +
geom_node_plot(gglist = list(
geom_bar(aes(x = "", fill = species), size = 0.2, width = 0.9,
position = position_fill(), color = "black"),
theme_minimal(base_family = "arial", base_size = 46),
scale_fill_manual(values = c("gray50","gray55","gray60",
"gray70","gray80","gray85",
"blue","gray95")),
xlab(""),
ylab("Probability"),
geom_text(aes(x = "", group = species,
label = stat(count)),
stat = "count", position = position_fill(),
vjust = 1., size = 12)),
shared_axis_labels = TRUE, size = 1.2)
#> Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
#> ℹ Please use `linewidth` instead.
#> This warning is displayed once every 8 hours.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was generated.
#> Warning: `stat(count)` was deprecated in ggplot2 3.4.0.
#> ℹ Please use `after_stat(count)` instead.
#> ℹ The deprecated feature was likely used in the ggparty package.
#> Please report the issue at <https://github.com/martin-borkovec/ggparty/issues>.
#> This warning is displayed once every 8 hours.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was generated.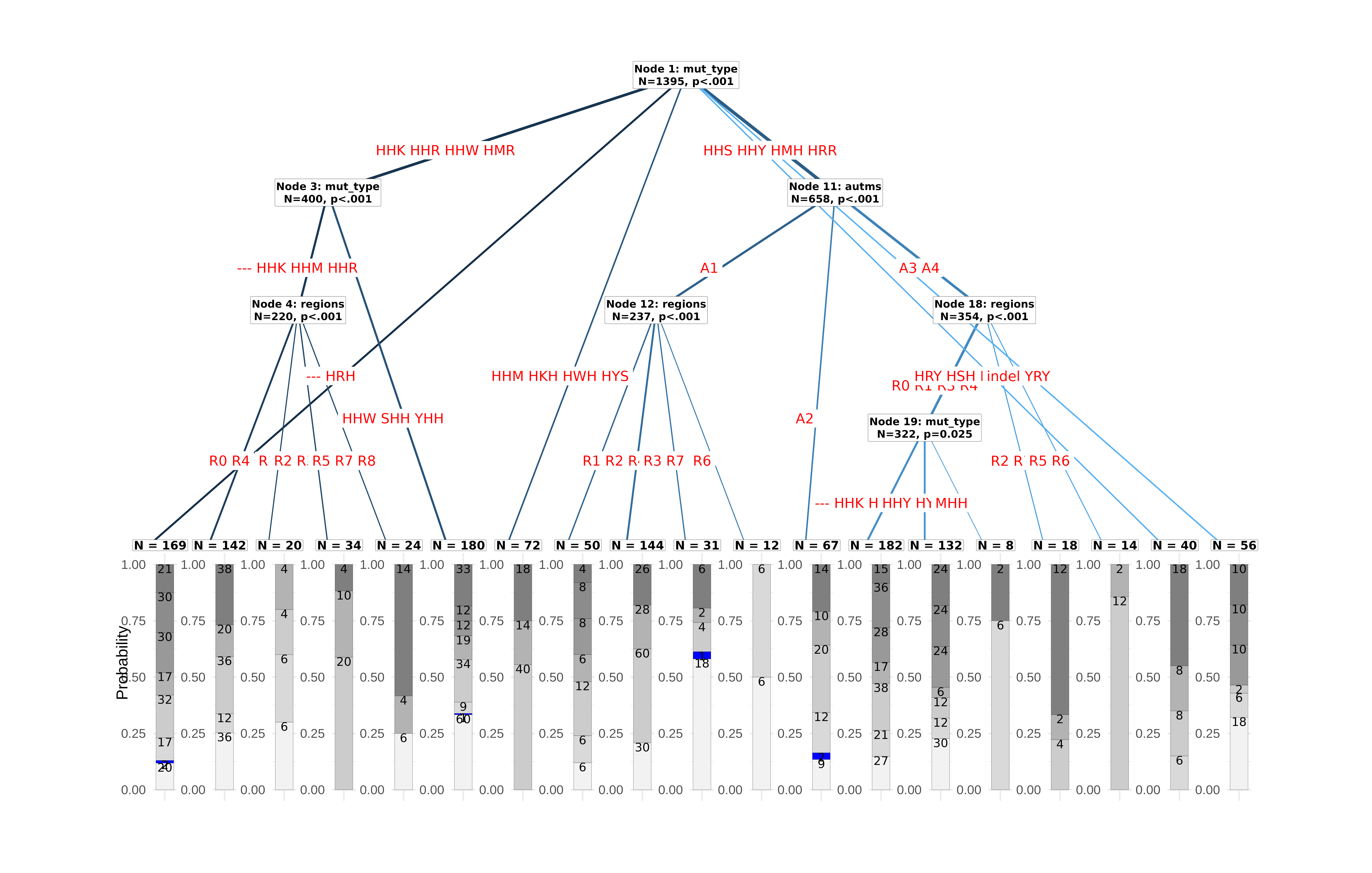
Stochastic-deterministic logical rules
Since only one mutational event human-to-human in region R1 from class A3 is reported in the right side of the tree, with high probability only non-humans hold the following rule:
rule <- (dat$autms == "A4" | (dat$autms == "A3" & dat$mut_type != "HRH"))
unique(as.character(dat[rule,]$species))
#> [1] "bolivian_monkey" "golden_monkey" "gelada_baboon" "silvery_gibbon" "chimpanzee" "bonobos"
#> [7] "gorilla"Only humans-to-human mutations hold the following rule:
idx <- dat$autm == "A1" & dat$regions == "R3" & (dat$mut_type == "HHY" | dat$mut_type == "YHH")
dat[ idx, ]
#> autms regions mut_type cube species
#> 632 A1 R3 YHH ACGT human
#> 687 A1 R3 HHY ACGT humanOnly non-humans hold the following rule
rule <- (dat$autms == "A4" | (dat$autms == "A3" & dat$regions != "R1"))
unique(as.character(dat[rule,]$species))
#> [1] "bolivian_monkey" "golden_monkey" "gelada_baboon" "silvery_gibbon" "gorilla" "chimpanzee"
#> [7] "bonobos"A printing version in TIFF format could be:
# tiff(filename = "~/home/chaid_tree.tiff",
# width = 3000, height = 1800, units = "px", pointsize = 1,
# compression = "lzw", bg = "white", res = 600)
#
# ggparty(chaid_tree) +
# geom_edge(aes(color = id), show.legend = FALSE) +
# geom_edge_label(size = 1, colour = "red", fontface = "bold") +
# geom_node_label(line_list = list(aes(label = paste0("Node ", id,": ", splitvar)),
# aes(label = paste0("N=", node.size[id], ", p",
# ifelse(pvals < .001, "<.001",
# paste0("=", round(pvals, 3)))))),
# line_gpar = list(list(size = 4),
# list(size = 4)),
# ids = "inner", label.size = 0.1) +
# geom_node_info(size = 2) +
# geom_node_label(aes(label = paste0("N = ", nodesize)),
# ids = "terminal", nudge_y = -0.0,
# nudge_x = 0.01, label.size = 0.1,
# line_gpar = list(list(size = 3.5),
# list(size = 3.5))) +
# geom_node_plot(gglist = list(
# geom_bar(aes(x = "", fill = species), size = 0.2, width = 10,
# position = position_fill(), color = "black"),
# theme_minimal(base_family = "arial", base_size = 5),
# scale_fill_manual(values = c("gray50","gray55","gray60",
# "gray70","gray80","gray85",
# "blue","gray95")),
# xlab(""),
# ylab("Probability"),
# geom_text(aes(x = "", group = species,
# label = stat(count)),
# stat = "count", position = position_fill(),
# vjust = 1., size = 1)),
# shared_axis_labels = TRUE, size = 1.)
#
#
# dev.off()Decision tree with a larger dataset
The analyses accomplished here (with a small dataset) are only for the purpose to illustrate the application of the theory, since for the sake of visualization and simplicity, were limited to small sample data set and to the application of a relatively “modest” (unsupervised) machine-learning approach which, however, is sufficient to illustrate the concepts. Next, the DNA sequence alignment has been increases adding 20 human sequences. Results, will obviously vary, since we will more valuable information about the human population. However, still the sample size for the rest of primates are pretty small and, by construction, the model predicting capability is strongly conditioned to the sample size.
The dataset is included with the package
data(brca1_aln2, package = "GenomAutomorphism")
brca1_aln2
#> DNAMultipleAlignment with 41 rows and 2283 columns
#> aln names
#> [1] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA NM_007298.3:20-22...
#> [2] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA U64805.1:1-2280_H...
#> [3] ATGGATTTCTCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA NM_007298.3:20-22...
#> [4] ATGGATTTGTCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA NM_007298.3:20-22...
#> [5] ATGGATTTTTCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA NM_007298.3:20-22...
#> [6] ATGGATTTTTCTGCTCTTCGGGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA NM_007298.3:20-22...
#> [7] ATGGATTTTTCTGCTCTTCGCTTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA NM_007298.3:20-22...
#> [8] ATGGATTTTTCTGCTCTTCGCCTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA NM_007298.3:20-22...
#> [9] ATGGATTTTTCTGCTCTTCGCATTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA NM_007298.3:20-22...
#> ... ...
#> [33] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCTCAGATCCCCCACAGCCACTACTGA XM_034941185.1:24...
#> [34] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCTCAGATCCCCCACAGCCACTACTGA XM_034941182.1:25...
#> [35] ATGGATTTATCTGCTGTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGATACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA XM_032163757.1:14...
#> [36] ATGGATTTATCTGCTGTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGATACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA XM_032163756.1:14...
#> [37] ATGGATTTATCTGCTGTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGATACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA XM_032163758.1:13...
#> [38] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACGCCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA XM_030923119.1:18...
#> [39] ATGGATTTATCTGCTCTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACGCCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA XM_030923118.1:18...
#> [40] ATGGATTTACCTGCTGTTCGCGTTGAAGAAGTACAAAATGTCATT...CTGGACACCTACCTGATACCCCAGATCCCCCACAGCCACTACTGA XM_025363316.1:14...
#> [41] ATGGATTTATCTGCTGTTCGTGTTGAAGAAGTGCAAAATGTCCTT...CTGGACACCTACCTGATACCCCAGATCCCTCACAGCCACTACTGA XM_039475995.1:49...The sample names
strtrim(names(brca1_aln2@unmasked), 100)
#> [1] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_DNA_repair_associated_(BRCA1)_transcript_variant_4_mRNA"
#> [2] "U64805.1:1-2280_Homo_sapiens_Brca1-delta11b_(Brca1)_mRNA_complete_cds"
#> [3] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_9A-C_L3F_rs780157871"
#> [4] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_9A-G_rs780157871"
#> [5] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_9A-T_L3F_rs780157871"
#> [6] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_21C-G_rs149402012"
#> [7] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_22G-T_V8F_rs528902306"
#> [8] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_22G-C_V8L_rs528902306"
#> [9] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_22G-A_V8I_rs528902306"
#> [10] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_75C-T_rs80356839"
#> [11] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_999T-A_rs1060915"
#> [12] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_985A-C_I329L_rs80357157"
#> [13] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_1014C-A_D338E_rs1057523168"
#> [14] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_1134A-C_rs1555582589."
#> [15] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_1327G-T_D443Y_rs28897691"
#> [16] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_1327G-C_D443H_rs28897691"
#> [17] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_1327G-A_D443N_rs28897691"
#> [18] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_1348T-A_L450M_rs80357431"
#> [19] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_2100T-C_rs2050995166"
#> [20] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_2102T-G_V701G_rs80356920"
#> [21] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_2102T-C_V701A_rs80356920"
#> [22] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_2102T-A_V701D_rs80356920"
#> [23] "NM_007298.3:20-2299_Homo_sapiens_BRCA1_transcript_variant_4_mRNA_305C-G_A102G_rs80357190"
#> [24] "XM_031011560.1:233-2515_PREDICTED:_Gorilla_gorilla_gorilla_BRCA1_DNA_repair_associated_(BRCA1)_trans"
#> [25] "XM_031011561.1:233-2512_PREDICTED:_Gorilla_gorilla_gorilla_BRCA1_DNA_repair_associated_(BRCA1)_trans"
#> [26] "XM_031011562.1:163-2442_PREDICTED:_Gorilla_gorilla_gorilla_BRCA1_DNA_repair_associated_(BRCA1)_trans"
#> [27] "XM_009432101.3:276-2555_PREDICTED:_Pan_troglodytes_BRCA1_DNA_repair_associated_(BRCA1)_transcript_va"
#> [28] "XM_009432104.3:371-2650_PREDICTED:_Pan_troglodytes_BRCA1_DNA_repair_associated_(BRCA1)_transcript_va"
#> [29] "XM_016930487.2:371-2650_PREDICTED:_Pan_troglodytes_BRCA1_DNA_repair_associated_(BRCA1)_transcript_va"
#> [30] "XM_009432099.3:371-2653_PREDICTED:_Pan_troglodytes_BRCA1_DNA_repair_associated_(BRCA1)_transcript_va"
#> [31] "XM_034941183.1:254-2533_PREDICTED:_Pan_paniscus_BRCA1_DNA_repair_associated_(BRCA1)_transcript_varia"
#> [32] "XM_034941184.1:254-2533_PREDICTED:_Pan_paniscus_BRCA1_DNA_repair_associated_(BRCA1)_transcript_varia"
#> [33] "XM_034941185.1:248-2527_PREDICTED:_Pan_paniscus_BRCA1_DNA_repair_associated_(BRCA1)_transcript_varia"
#> [34] "XM_034941182.1:254-2536_PREDICTED:_Pan_paniscus_BRCA1_DNA_repair_associated_(BRCA1)_transcript_varia"
#> [35] "XM_032163757.1:145-2418_PREDICTED:_Hylobates_moloch_BRCA1_DNA_repair_associated_(BRCA1)_transcript_v"
#> [36] "XM_032163756.1:145-2421_PREDICTED:_Hylobates_moloch_BRCA1_DNA_repair_associated_(BRCA1)_transcript_v"
#> [37] "XM_032163758.1:139-2412_PREDICTED:_Hylobates_moloch_BRCA1_DNA_repair_associated_(BRCA1)_transcript_v"
#> [38] "XM_030923119.1:184-2463_PREDICTED:_Rhinopithecus_roxellana_BRCA1_DNA_repair_associated_(BRCA1)_trans"
#> [39] "XM_030923118.1:183-2465_PREDICTED:_Rhinopithecus_roxellana_BRCA1_DNA_repair_associated_(BRCA1)_trans"
#> [40] "XM_025363316.1:147-2426_PREDICTED:_Theropithecus_gelada_BRCA1_DNA_repair_associated_(BRCA1)_transcri"
#> [41] "XM_039475995.1:49-2328_PREDICTED:_Saimiri_boliviensis_boliviensis_BRCA1_DNA_repair_associated_(BRCA1"The automorphisms can be computed as before. For the sake of saving time object brca1_autm2 is included with package
## Do not run it. This is included with package
nams <- c(paste0("human_1.", 0:21),"human_2","gorilla_1","gorilla_2","gorilla_3",
"chimpanzee_1","chimpanzee_2","chimpanzee_3","chimpanzee_4",
"bonobos_1","bonobos_2","bonobos_3","bonobos_4","silvery_gibbon_1",
"silvery_gibbon_1","silvery_gibbon_3","golden_monkey_1",
"golden_monkey_2","gelada_baboon","bolivian_monkey")
brca1_autm2 <- automorphisms(
seqs = brca1_aln2,
group = "Z64",
cube = c("ACGT", "TGCA"),
cube_alt = c("CATG", "GTAC"),
nms = nams,
verbose = FALSE)Object brca1_autm2 can be load typing:
data(brca1_autm2, package = "GenomAutomorphism")
brca1_autm2
#> AutomorphismList object of length: 820
#> names(820): human_1.0.human_1.1 human_1.0.human_1.2 human_1.0.human_1.3 ... golden_monkey_2.gelada_baboon golden_monkey_2.bolivian_monkey gelada_baboon.bolivian_monkey
#> -------
#> Automorphism object with 761 ranges and 8 metadata columns:
#> seqnames ranges strand | seq1 seq2 aa1 aa2 coord1 coord2 autm
#> <Rle> <IRanges> <Rle> | <character> <character> <character> <character> <numeric> <numeric> <numeric>
#> [1] 1 1 + | ATG ATG M M 50 50 1
#> [2] 1 2 + | GAT GAT D D 11 11 1
#> [3] 1 3 + | TTA TTA L L 60 60 1
#> [4] 1 4 + | TCT TCT S S 31 31 1
#> [5] 1 5 + | GCT GCT A A 27 27 1
#> ... ... ... ... . ... ... ... ... ... ... ...
#> [757] 1 757 + | CAC CAC H H 5 5 1
#> [758] 1 758 + | AGC AGC S S 33 33 1
#> [759] 1 759 + | CAC CAC H H 5 5 1
#> [760] 1 760 + | TAC TAC Y Y 13 13 1
#> [761] 1 761 + | TGA TGA * * 44 44 1
#> cube
#> <character>
#> [1] ACGT
#> [2] ACGT
#> [3] ACGT
#> [4] ACGT
#> [5] ACGT
#> ... ...
#> [757] ACGT
#> [758] ACGT
#> [759] ACGT
#> [760] ACGT
#> [761] ACGT
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths
#> ...
#> <819 more DFrame element(s)>
#> Two slots: 'DataList' & 'SeqRanges'
#> -------As before we are interested on mutational events in respect to human (as reference).
nams <- names(brca1_autm2)
idx1 <- grep("human_1.", nams)
idx2 <- grep("human_2.", nams)
idx <- union(idx1, idx2)
h_brca1_autm <- unlist(brca1_autm2[ idx ])
h_brca1_autm = h_brca1_autm[ which(h_brca1_autm$autm != 1) ]
h_brca1_autm
#> Automorphism object with 16542 ranges and 8 metadata columns:
#> seqnames ranges strand | seq1 seq2 aa1 aa2 coord1
#> <Rle> <IRanges> <Rle> | <character> <character> <character> <character> <numeric>
#> human_1.0.human_1.1 1 239 + | CAT CGT H R 7
#> human_1.0.human_1.1 1 253 + | GCA GTA A V 24
#> human_1.0.human_1.1 1 323 + | TCT CCT S P 31
#> human_1.0.human_1.1 1 333 + | TCT TCC S S 31
#> human_1.0.human_1.1 1 350 + | --- --- - - NA
#> ... ... ... ... . ... ... ... ... ...
#> human_2.bolivian_monkey 1 716 + | AAT AGT N S 3
#> human_2.bolivian_monkey 1 726 + | GAG GAA E E 10
#> human_2.bolivian_monkey 1 730 + | GTG GTA V V 58
#> human_2.bolivian_monkey 1 731 + | ACC ACT T T 17
#> human_2.bolivian_monkey 1 756 + | CCC CCT P P 21
#> coord2 autm cube
#> <numeric> <numeric> <character>
#> human_1.0.human_1.1 39 33 ACGT
#> human_1.0.human_1.1 56 21 ACGT
#> human_1.0.human_1.1 23 9 ACGT
#> human_1.0.human_1.1 29 3 ACGT
#> human_1.0.human_1.1 NA -1 Gaps
#> ... ... ... ...
#> human_2.bolivian_monkey 35 33 ACGT
#> human_2.bolivian_monkey 8 52 ACGT
#> human_2.bolivian_monkey 56 12 ACGT
#> human_2.bolivian_monkey 19 35 ACGT
#> human_2.bolivian_monkey 23 59 ACGT
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengthsBar plot automorphism distribution by coefficient (II)
The automorphism distribution by cubes can be summarized in the bar-plot graphic.
Object autby_coef carried all the pairwise comparisons, while it will be enough to use data from a single species as reference, e.g., humans.
First the data must be reordered into a object:
h_autby_coef <- automorphism_bycoef(x = h_brca1_autm)
h_autby_coef
#> AutomorphismByCoef object with 16515 ranges and 7 metadata columns:
#> seqnames ranges strand | seq1 seq2 aa1 aa2 autm
#> <Rle> <IRanges> <Rle> | <character> <character> <character> <character> <numeric>
#> human_1.0.human_1.2 1 3 + | TTA TTC L F 22
#> human_1.0.human_1.3 1 3 + | TTA TTG L L 43
#> human_1.0.human_1.4 1 3 + | TTA TTT L F 0
#> human_1.0.human_1.5 1 3 + | TTA TTT L F 0
#> human_1.0.human_1.6 1 3 + | TTA TTT L F 0
#> ... ... ... ... . ... ... ... ... ...
#> human_1.18.bolivian_monkey 1 756 + | CCC CCT P P 59
#> human_1.19.bolivian_monkey 1 756 + | CCC CCT P P 59
#> human_1.20.bolivian_monkey 1 756 + | CCC CCT P P 59
#> human_1.21.bolivian_monkey 1 756 + | CCC CCT P P 59
#> human_2.bolivian_monkey 1 756 + | CCC CCT P P 59
#> mut_type cube
#> <character> <character>
#> human_1.0.human_1.2 HHM TGCA
#> human_1.0.human_1.3 HHR TGCA
#> human_1.0.human_1.4 HHW Trnl
#> human_1.0.human_1.5 HHW Trnl
#> human_1.0.human_1.6 HHW Trnl
#> ... ... ...
#> human_1.18.bolivian_monkey HHY ACGT
#> human_1.19.bolivian_monkey HHY ACGT
#> human_1.20.bolivian_monkey HHY ACGT
#> human_1.21.bolivian_monkey HHY ACGT
#> human_2.bolivian_monkey HHY ACGT
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengthsThe counts by type of mutations
table(h_autby_coef$mut_type)
#>
#> --- HHH HHK HHM HHR HHS HHW HHY HKH HMH HMR HRH HRR HRW HRY HSH HWH HYH HYS KHH KKH KWH KYH MHH
#> 575 391 290 447 2050 270 769 2358 126 829 253 1648 69 3 46 271 195 626 46 179 3 3 3 573
#> RHH RKH RMH RWH RYH SHH SHM WHH WHK YHH YRH YRY
#> 2469 1 46 1 1 874 1 229 23 755 69 23Simplifying species names
nams <- names(h_autby_coef)
nams <- sub("human[_][1-2][.]", "", nams)
nams <- sub("[_][1-6]", "", nams)
nams <- sub("[0123456789]*[.]", "", nams)
nams <- sub("[.][0123456789]*", "", nams)
dt <- data.frame(h_autby_coef, species = nams)
dt <- data.frame(dt, species = nams)
dt <- dt[, c("start", "autm", "species", "mut_type", "aa1", "aa2", "cube")]
DataFrame(dt)
#> DataFrame with 16515 rows and 7 columns
#> start autm species mut_type aa1 aa2 cube
#> <integer> <numeric> <character> <character> <character> <character> <character>
#> 1 3 22 human HHM L F TGCA
#> 2 3 43 human HHR L L TGCA
#> 3 3 0 human HHW L F Trnl
#> 4 3 0 human HHW L F Trnl
#> 5 3 0 human HHW L F Trnl
#> ... ... ... ... ... ... ... ...
#> 16511 756 59 bolivian_monkey HHY P P ACGT
#> 16512 756 59 bolivian_monkey HHY P P ACGT
#> 16513 756 59 bolivian_monkey HHY P P ACGT
#> 16514 756 59 bolivian_monkey HHY P P ACGT
#> 16515 756 59 bolivian_monkey HHY P P ACGTNominal variables are transformed into
dt$start <- as.numeric(dt$start)
dt$autm <- as.numeric(dt$autm)
dt$cube <- as.factor(dt$cube)
dt$species <- as.factor(dt$species)
dt$mut_type <- as.factor(dt$mut_type)
counts <- table(dt$cube)
par(family = "serif", cex = 0.6, font = 2, mar=c(4,6,4,4))
barplot(counts, #main="Automorphism distribution",
xlab="Genetic-code cube representation",
ylab="Fixed mutational events",
col=c("darkblue","red", "darkgreen", "magenta"),
border = NA, axes = F, #ylim = c(0, 6000),
cex.lab = 2, cex.main = 1.5, cex.names = 2)
axis(2, at = c(0, 2000, 4000, 6000, 8000, 10000, 12000), cex.axis = 1.5)
mtext(side = 1,line = -2.8, at = c(0.7, 1.9, 3.1, 4.3),
text = paste0( counts ), cex = 1.4,
col = c("white", "black","white", "red"))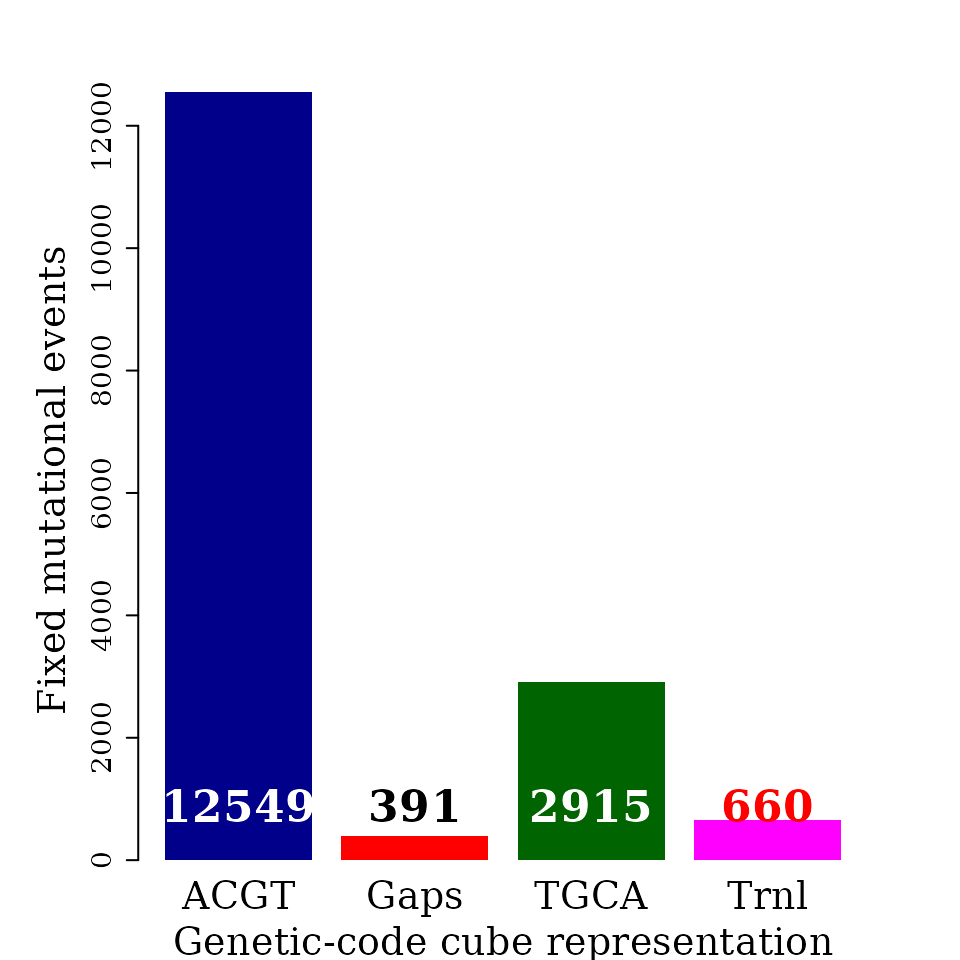
Classification Tree. Chi-squared Automated Interaction Detection
In this analysis a new variable has been include, which derives from the estimation of the contact potential matrix of amino acids made by Miyazawa and Jernigan (5), which are considered quasichemical energy of interactions in an average buried environment. The amino acids potentials, as well as, others numerical indices representing various physicochemical and biochemical properties of amino acids and pairs of amino acids can be found in AAindex.
The lists of available aminoacid similarity and statistical potential matrices can be found with functions: aaindex2 and aaindex3, respectively.
data("aaindex3", package = "GenomAutomorphism")
interval <- function(x, a, b) {
x >= a & x <= b
}
datos <- dt
datos$mut_cost <- get_mutscore(aa1 = datos$aa1, aa2 = datos$aa2,
acc = "MIYS960102", aaindex = "aaindex3", alphabet = "AA",
num.cores = 20)
hist(datos$mut_cost,12)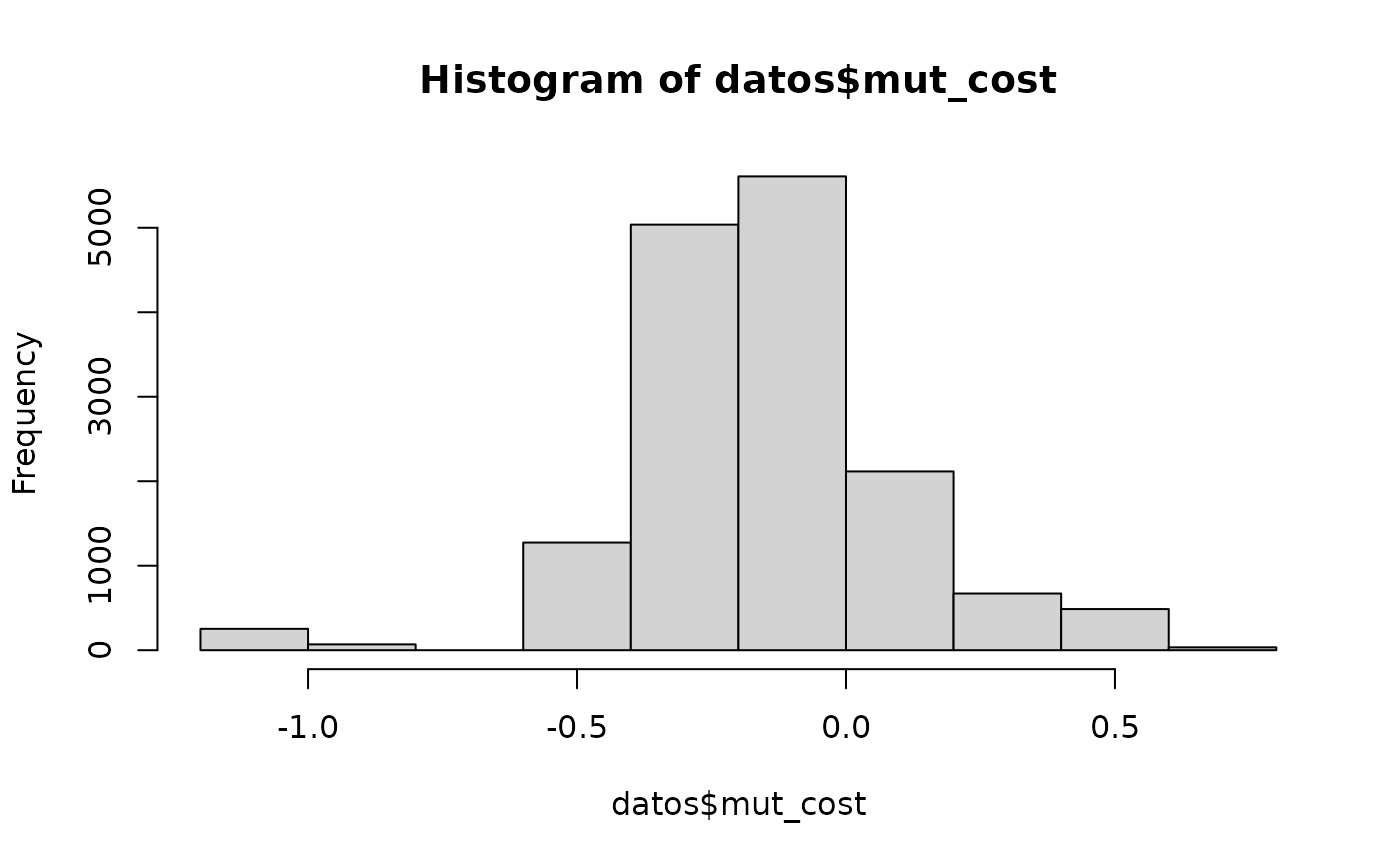
datos$mut_cost[ is.na(datos$mut_cost) ] <- -10
datos$autms <- case_when(datos$autm < 16 ~ 'A1',
interval(datos$autm, 16, 31) ~ 'A2',
interval(datos$autm, 32, 47) ~ 'A3',
datos$autm > 47 ~ 'A4')
datos$autms <- as.factor(datos$autms)
datos$mut_type <- as.character(datos$mut_type)
datos$mut_type <- as.factor(datos$mut_type)
datos$regions <- case_when(datos$start < 102 ~ 'R0',
interval(datos$start, 103, 229) ~ 'R0.',
interval(datos$start, 230, 270) ~ 'R1',
interval(datos$start, 271, 305) ~ 'R2',
interval(datos$start, 306, 338) ~ 'R3',
interval(datos$start, 339, 533) ~ 'R4',
interval(datos$start, 534, 570) ~ 'R5',
interval(datos$start, 571, 653) ~ 'R6',
interval(datos$start, 654, 709) ~ 'R7',
datos$start > 709 ~ 'R8')
datos$regions <- as.factor(datos$regions)
datos$autm <- as.factor(datos$autm)
datos$species <- as.factor(datos$species)
datos$start <- as.factor(datos$start)
datos$cube <- as.factor(datos$cube)
datos$mut_cost <- as.factor(datos$mut_cost)
datos <- datos[, c( "autms", "regions", "mut_type", "cube", "mut_cost", "species")]
DataFrame(datos)
#> DataFrame with 16515 rows and 6 columns
#> autms regions mut_type cube mut_cost species
#> <factor> <factor> <factor> <factor> <factor> <factor>
#> 1 A2 R0 HHM TGCA -0.26 human
#> 2 A3 R0 HHR TGCA -0.3 human
#> 3 A1 R0 HHW Trnl -0.26 human
#> 4 A1 R0 HHW Trnl -0.26 human
#> 5 A1 R0 HHW Trnl -0.26 human
#> ... ... ... ... ... ... ...
#> 16511 A4 R8 HHY ACGT -0.12 bolivian_monkey
#> 16512 A4 R8 HHY ACGT -0.12 bolivian_monkey
#> 16513 A4 R8 HHY ACGT -0.12 bolivian_monkey
#> 16514 A4 R8 HHY ACGT -0.12 bolivian_monkey
#> 16515 A4 R8 HHY ACGT -0.12 bolivian_monkeyA classification tree is estimated with CHAID algorithm:
ctrl <- chaid_control(minsplit = 500, minprob = 0.9, alpha2 = 0.01, alpha4 = 0.01)
chaid_res <- chaid(species ~ autms + regions + mut_type + cube + mut_cost , data = datos,
control = ctrl)
chaid_res
#>
#> Model formula:
#> species ~ autms + regions + mut_type + cube + mut_cost
#>
#> Fitted party:
#> [1] root
#> | [2] mut_cost in -10
#> | | [3] mut_type in ---, HHK, HHM, HHR, HHS, HHW, HHY, HKH, HMH, HMR, HRH, HRR, HRW, HRY, HSH, HWH, HYH, HYS, KHH, KKH, KWH, KYH, MHH, RHH, RKH, RMH, RWH, RYH, SHH, SHM, WHH, WHK, YHH, YRH, YRY
#> | | | [4] regions in R0, R0., R1, R3, R5, R6, R7, R8: silvery_gibbon (n = 253, err = 36.4%)
#> | | | [5] regions in R2: bonobos (n = 23, err = 0.0%)
#> | | | [6] regions in R4: bonobos (n = 299, err = 76.9%)
#> | | [7] mut_type in HHH: human (n = 391, err = 35.3%)
#> | [8] mut_cost in -1.04, -0.02, 0.1, 0.13, 0.14, 0.16, 0.23
#> | | [9] mut_cost in -10, -1.04, -0.85, -0.48, -0.47, -0.41, -0.39, -0.36, -0.33, -0.32, -0.3, -0.29, -0.26, -0.24, -0.23, -0.22, -0.21, -0.2, -0.19, -0.18, -0.15, -0.14, -0.13, -0.12, -0.11, -0.1, -0.09, -0.08, -0.07, -0.06, -0.05, -0.04, -0.03, -0.02, -0.01, 0.02, 0.03, 0.06, 0.08, 0.1, 0.12, 0.13, 0.14, 0.15, 0.17, 0.21, 0.22, 0.27, 0.28, 0.3, 0.34, 0.37, 0.46, 0.49, 0.54, 0.57, 0.7
#> | | | [10] mut_type in ---, HHH, HHK, HHM, HHR, HHS, HHW, HHY, HKH, HMH, HMR, HRH, HRR, HRW, HRY, HWH, HYH, HYS, KHH, KKH, KWH, KYH, MHH, RHH, RKH, RMH, RWH, RYH, SHH, SHM, WHK, YHH, YRH
#> | | | | [11] regions in R0, R1, R2, R3, R6, R7, R8: golden_monkey (n = 69, err = 33.3%)
#> | | | | [12] regions in R0.: golden_monkey (n = 207, err = 55.6%)
#> | | | | [13] regions in R4: golden_monkey (n = 322, err = 42.9%)
#> | | | | [14] regions in R5: gelada_baboon (n = 92, err = 50.0%)
#> | | | [15] mut_type in HSH, YRY: bolivian_monkey (n = 46, err = 0.0%)
#> | | | [16] mut_type in WHH: golden_monkey (n = 69, err = 33.3%)
#> | | [17] mut_cost in 0.16, 0.23: bolivian_monkey (n = 24, err = 4.2%)
#> | [18] mut_cost in -0.85, -0.48, -0.14, 0.02, 0.28: bolivian_monkey (n = 121, err = 41.3%)
#> | [19] mut_cost in -0.47
#> | | [20] regions in R0, R0., R3, R5, R6, R7, R8: bonobos (n = 92, err = 0.0%)
#> | | [21] regions in R1: bolivian_monkey (n = 23, err = 0.0%)
#> | | [22] regions in R2: bonobos (n = 414, err = 77.8%)
#> | | [23] regions in R4
#> | | | [24] autms in A1, A2: human (n = 42, err = 71.4%)
#> | | | [25] autms in A3: bonobos (n = 488, err = 81.1%)
#> | | | [26] autms in A4: human (n = 14, err = 0.0%)
#> | [27] mut_cost in -0.41: golden_monkey (n = 155, err = 69.0%)
#> | [28] mut_cost in -0.39: silvery_gibbon (n = 276, err = 50.0%)
#> | [29] mut_cost in -0.36: golden_monkey (n = 109, err = 56.0%)
#> | [30] mut_cost in -0.33: silvery_gibbon (n = 150, err = 54.0%)
#> | [31] mut_cost in -0.32, -0.18: bolivian_monkey (n = 414, err = 66.7%)
#> | [32] mut_cost in -0.3
#> | | [33] regions in R0, R1, R3, R8: golden_monkey (n = 164, err = 70.7%)
#> | | [34] regions in R0.: silvery_gibbon (n = 138, err = 50.0%)
#> | | [35] regions in R2: golden_monkey (n = 138, err = 33.3%)
#> | | [36] regions in R4: bolivian_monkey (n = 161, err = 42.9%)
#> | | [37] regions in R5: bolivian_monkey (n = 46, err = 0.0%)
#> | | [38] regions in R6: golden_monkey (n = 92, err = 50.0%)
#> | | [39] regions in R7: gelada_baboon (n = 23, err = 0.0%)
#> | [40] mut_cost in -0.29
#> | | [41] regions in R0: human (n = 74, err = 55.4%)
#> | | [42] regions in R0., R2, R5, R8: bolivian_monkey (n = 92, err = 0.0%)
#> | | [43] regions in R1: golden_monkey (n = 92, err = 50.0%)
#> | | [44] regions in R3: silvery_gibbon (n = 137, err = 49.6%)
#> | | [45] regions in R4: golden_monkey (n = 299, err = 53.8%)
#> | | [46] regions in R6: silvery_gibbon (n = 138, err = 50.0%)
#> | | [47] regions in R7: bolivian_monkey (n = 135, err = 34.1%)
#> | [48] mut_cost in -0.26
#> | | [49] regions in R0, R6, R8: human (n = 379, err = 66.5%)
#> | | [50] regions in R0.
#> | | | [51] autms in A1, A2: bonobos (n = 437, err = 78.9%)
#> | | | [52] autms in A3: silvery_gibbon (n = 69, err = 0.0%)
#> | | | [53] autms in A4: golden_monkey (n = 46, err = 0.0%)
#> | | [54] regions in R1: golden_monkey (n = 69, err = 33.3%)
#> | | [55] regions in R2: golden_monkey (n = 92, err = 50.0%)
#> | | [56] regions in R3: human (n = 79, err = 45.6%)
#> | | [57] regions in R4, R7: golden_monkey (n = 391, err = 52.9%)
#> | | [58] regions in R5: bolivian_monkey (n = 23, err = 0.0%)
#> | [59] mut_cost in -0.24, -0.03: bolivian_monkey (n = 443, err = 79.2%)
#> | [60] mut_cost in -0.23: golden_monkey (n = 339, err = 72.3%)
#> | [61] mut_cost in -0.22: golden_monkey (n = 69, err = 33.3%)
#> | [62] mut_cost in -0.21: gelada_baboon (n = 24, err = 4.2%)
#> | [63] mut_cost in -0.2: gorilla (n = 109, err = 33.9%)
#> | [64] mut_cost in -0.19: golden_monkey (n = 420, err = 57.1%)
#> | [65] mut_cost in -0.15: gorilla (n = 188, err = 63.3%)
#> | [66] mut_cost in -0.13, 0.57: silvery_gibbon (n = 254, err = 69.3%)
#> | [67] mut_cost in -0.12, 0.22
#> | | [68] regions in R0, R1, R2, R3, R5: golden_monkey (n = 128, err = 64.1%)
#> | | [69] regions in R0.: bonobos (n = 184, err = 50.0%)
#> | | [70] regions in R4: gelada_baboon (n = 23, err = 0.0%)
#> | | [71] regions in R6: bolivian_monkey (n = 23, err = 0.0%)
#> | | [72] regions in R7, R8: bonobos (n = 276, err = 66.7%)
#> | [73] mut_cost in -0.11: bolivian_monkey (n = 24, err = 4.2%)
#> | [74] mut_cost in -0.1: golden_monkey (n = 71, err = 35.2%)
#> | [75] mut_cost in -0.09, 0.17
#> | | [76] regions in R0, R3, R5, R7: gorilla (n = 184, err = 62.5%)
#> | | [77] regions in R0.: silvery_gibbon (n = 276, err = 50.0%)
#> | | [78] regions in R1: gorilla (n = 161, err = 57.1%)
#> | | [79] regions in R2: golden_monkey (n = 46, err = 0.0%)
#> | | [80] regions in R4: golden_monkey (n = 368, err = 62.5%)
#> | | [81] regions in R6: golden_monkey (n = 69, err = 33.3%)
#> | | [82] regions in R8: bolivian_monkey (n = 23, err = 0.0%)
#> | [83] mut_cost in -0.08: silvery_gibbon (n = 92, err = 25.0%)
#> | [84] mut_cost in -0.07: golden_monkey (n = 230, err = 60.0%)
#> | [85] mut_cost in -0.06: golden_monkey (n = 219, err = 58.0%)
#> | [86] mut_cost in -0.05, 0.54
#> | | [87] regions in R0, R2, R5, R6, R8: gelada_baboon (n = 23, err = 0.0%)
#> | | [88] regions in R0., R7: bonobos (n = 392, err = 76.5%)
#> | | [89] regions in R1: golden_monkey (n = 253, err = 63.6%)
#> | | [90] regions in R3: silvery_gibbon (n = 109, err = 33.9%)
#> | | [91] regions in R4: silvery_gibbon (n = 92, err = 25.0%)
#> | [92] mut_cost in -0.04, 0.06, 0.34, 0.46
#> | | [93] regions in R0, R5, R6, R8: golden_monkey (n = 92, err = 50.0%)
#> | | [94] regions in R0.: bolivian_monkey (n = 69, err = 0.0%)
#> | | [95] regions in R1: golden_monkey (n = 129, err = 62.8%)
#> | | [96] regions in R2: golden_monkey (n = 69, err = 33.3%)
#> | | [97] regions in R3: golden_monkey (n = 109, err = 56.0%)
#> | | [98] regions in R4
#> | | | [99] mut_type in ---, HHH, HHK, HHM, HHR, HHS, HHY, HKH, HMH, HMR, HRR, HRW, HRY, HSH, HWH, HYH, HYS, KHH, KKH, KWH, KYH, MHH, RHH, RKH, RMH, RWH, RYH, SHH, SHM, WHH, WHK, YHH, YRH, YRY: golden_monkey (n = 69, err = 33.3%)
#> | | | [100] mut_type in HHW: bolivian_monkey (n = 23, err = 0.0%)
#> | | | [101] mut_type in HRH: bonobos (n = 414, err = 77.8%)
#> | | [102] regions in R7: bolivian_monkey (n = 23, err = 0.0%)
#> | [103] mut_cost in -0.01
#> | | [104] regions in R0, R0., R2, R3, R7: bonobos (n = 88, err = 0.0%)
#> | | [105] regions in R1: chimpanzee (n = 326, err = 71.8%)
#> | | [106] regions in R4, R5: bolivian_monkey (n = 46, err = 0.0%)
#> | | [107] regions in R6: golden_monkey (n = 69, err = 33.3%)
#> | | [108] regions in R8: golden_monkey (n = 46, err = 0.0%)
#> | [109] mut_cost in 0.03: silvery_gibbon (n = 388, err = 65.2%)
#> | [110] mut_cost in 0.08: bolivian_monkey (n = 69, err = 0.0%)
#> | [111] mut_cost in 0.12
#> | | [112] autms in A1, A2: silvery_gibbon (n = 184, err = 62.5%)
#> | | [113] autms in A3: human (n = 39, err = 43.6%)
#> | | [114] autms in A4: bolivian_monkey (n = 299, err = 53.8%)
#> | [115] mut_cost in 0.15: golden_monkey (n = 333, err = 58.6%)
#> | [116] mut_cost in 0.21: golden_monkey (n = 46, err = 0.0%)
#> | [117] mut_cost in 0.27: bolivian_monkey (n = 210, err = 67.1%)
#> | [118] mut_cost in 0.3: silvery_gibbon (n = 207, err = 0.0%)
#> | [119] mut_cost in 0.37, 0.7: silvery_gibbon (n = 172, err = 58.1%)
#> | [120] mut_cost in 0.49: bonobos (n = 437, err = 78.9%)
#>
#> Number of inner nodes: 19
#> Number of terminal nodes: 101Plotting the CHAID tree (II)
Next, the data must be prepared for plotting the tree with ggparty:
## Updating CHAID decision tree
dp <- data_party(chaid_res)
dat <- dp[, c("autms", "regions", "mut_type", "cube", "mut_cost")]
dat$species <- dp[, "(response)"]
chaid_tree <- party(node = node_party(chaid_res),
data = dat,
fitted = dp[, c("(fitted)", "(response)")],
names = names(chaid_res))
## Extract p-values
pvals <- unlist(nodeapply(chaid_tree, ids = nodeids(chaid_tree), function(n) {
pvals <- info_node(n)$adjpvals
pvals < pvals[ which.min(pvals) ]
return(pvals)
}))
pvals <- pvals[ pvals < 0.05 ]
## Counts of event per spciees on each node
node.freq <- sapply(seq_along(chaid_tree), function(id) {
y <- data_party(chaid_tree, id = id)
y <- y[[ "(response)" ]]
table(y)
})
## total counts on each
node.size = colSums(node.freq)Plotting the tree with ggparty (font size adjusted for html output)
ggparty(chaid_tree, horizontal = TRUE) +
geom_edge(aes(color = id, size = node.size[id]/300), show.legend = FALSE) +
geom_edge_label(size = 12, colour = "red",
fontface = "bold",
shift = 0.64,
nudge_x = -0.01,
max_length = 10,
splitlevels = 1:4) +
geom_node_label(line_list = list(aes(label = paste0("Node ", id, ": ", splitvar)),
aes(label = paste0("N=", node.size[id], ", p",
ifelse(pvals < .001, "<.001",
paste0("=", round(pvals, 3)))),
size = 30)),
line_gpar = list(list(size = 30),
list(size = 30)),
ids = "inner", fontface = "bold", size = 30) +
geom_node_info() +
geom_node_label(aes(label = paste0("N = ", node.size),
fontface = "bold"),
ids = "terminal", nudge_y = -0.0, nudge_x = 0.01, size = 12) +
geom_node_plot(gglist = list(
geom_bar(aes(x = "", fill = species), size = 0.7, width = 0.9,
position = position_fill(), color = "black"),
theme_minimal(base_family = "arial", base_size = 20),
theme(legend.key.size = unit(2, "cm"),
legend.key.width = unit(3.5, 'cm'),
legend.text = element_text(size = 50),
legend.title = element_text(size = 52),
text = element_text(size = 44)),
coord_flip(),
scale_fill_manual(values = c("gray50","gray55","gray60",
"gray70","gray80","gray85",
"dodgerblue","gray95")),
xlab(""),
ylab("Probability"),
geom_text(aes(x = "", group = species,
label = stat(count)),
stat = "count", position = position_fill(),
vjust = 0.6, hjust = 1.5, size = 14)),
shared_axis_labels = TRUE, size = 1.4) 
It can immediately noticed that the quasichemical energy of interactions in an average buried environment suggest that the contact energy of amino acid buried in the protein inner would play a fundamental role in fixing mutational events in species populations.
Stochastic-deterministic logical rules
Since only one mutational event human-to-human in region R1 from class A3 is reported in the right side of the tree, with high probability only non-humans hold the following rule:
rule <- (dat$mut_cost == 0.03 )
table(as.character(dat[rule,]$species))
#>
#> bolivian_monkey gelada_baboon golden_monkey gorilla silvery_gibbon
#> 46 46 92 69 135Only humans-to-human mutations hold the following rule:
rule <- (dat$mut_cost == -0.47 & dat$regions == "R4" & dat$autm == "A4")
table(as.character(dat[ rule, ]$species))
#>
#> human
#> 14Only bonobos hold the following rule
rule <- (dat$mut_cost == -0.47 & is.element(dat$regions, c("R0","R0.","R3","R5")))
table(as.character(dat[rule,]$species))
#>
#> bonobos
#> 92Only Bolivian monkey holds:
rule <- (dat$mut_cost == 0.08 )
table(as.character(dat[rule,]$species))
#>
#> bolivian_monkey
#> 69References
1. Sanchez R, Morgado E, Grau R. Gene algebra from a genetic code algebraic structure. J Math Biol. 2005 Oct;51(4):431-57. doi: 10.1007/s00285-005-0332-8. Epub 2005 Jul 13. PMID: 16012800. ( PDF).
2. Robersy Sanchez, Jesús Barreto (2021) Genomic Abelian Finite Groups. doi: 10.1101/2021.06.01.446543.
3. M. V José, E.R. Morgado, R. Sánchez, T. Govezensky, The 24 possible algebraic representations of the standard genetic code in six or in three dimensions, Adv. Stud. Biol. 4 (2012) 119–152.PDF.
4. R. Sanchez. Symmetric Group of the Genetic–Code Cubes. Effect of the Genetic–Code Architecture on the Evolutionary Process MATCH Commun. Math. Comput. Chem. 79 (2018) 527-560. PDF.
5. Miyazawa, S. and Jernigan, R.L. Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J. Mol. Biol. 256, 623-644 (1996). PDF
6. Kawashima, S., Pokarowski, P., Pokarowska, M., Kolinski, A., Katayama, T., and Kanehisa, M.; AAindex: amino acid index database, progress report 2008. Nucleic Acids Res. 36, D202-D205 (2008). PMID:17998252. PDF